
Primul tratament specific pentru boala Batten, o afecțiune genetică rară greu de diagnosticat
95% dintre bolile rare nu au nici macar un tratament aprobat de FDA. În cele 5% dintre cazurile unde există un tratament, pacienții se luptă să aibă acces la el din cauza costurilor ridicate. Ceroid lipofuscinoza neuronală de tip 2 (CLN2) este o boală neurodegenerativă rară care afectează copii de 2-4 ani, caracterizată printr-o speranță de viață scăzută, pentru care FDA a aprobat anul acesta primul tratament, medicamentul orfan Brineura.
Ceroid lipofuscinozele neuronale reprezintă un grup de boli de stocaj lizozomal, cunoscute de obicei sub numele de boala Batten (deși există autori care susțin că boala Batten se referă specific la CLN tip 3). Aceastea se caracterizează prin mutații genetice care rezultă în deficite enzimatice la nivelul lizozomilor, componentele celulei responsabile de procesele de digestie. Astfel, materialele reziduale nu se mai pot elimina și se acumulează sub forma unui agregat de grăsimi – lipofuscina, numită și pigment de uzură.

Concentrarea pigmentului în special la nivelul cortexul cerebral și cerebelos determină simptomele acestei afecțiuni, simptome care pretează adesea la confuzii cu alte boli, ceea ce întârzie diagnosticul:
- tulburări de vedere care pot merge până la orbire
- crize epileptice
- deficit motor progresiv, dificultatea coordonării mișcărilor (ataxie)
- tulburări cognitive care pot merge până la demență
- tulburări de limbaj.
Brineura (cerliponaza alfa), obținută prin tehnici de recombinare genetică, înlocuiește enzima deficitară în această boală – TPP1 (tripeptidyl peptidase 1). FDA a aprobat medicamentul în aprilie 2017, iar în iunie acest medicament orfan a devenit disponibil și în Europa. Substanța se administrează direct la nivelul ventriculilor cerebrali (cavitățile pline cu lichid ale creierului) printr-un dispozitiv implantat chirugical.
Jean Jeacques Bienaimé, directorul companiei BioMarin, care produce Brineura, a declarat:
„Ne concentrăm în prezent eforturile asupra reducerii timpului până la obținerea diagnosticului corect. Pacienții, de obicei, nu sunt diagnosticați până la vârsta de 5 ani, adică la aproape 2 ani de la prima criză epileptică. Cu tratamentul care este acum disponibil, accelerarea procesului de diagnosticare va fi crucială”
În medie, un pacient cu o boală rară este examinat de 8 medici și primește 2-3 diagnostice greșite până la identificarea bolii. Timpul mediu până la diagnosticul corect este de 7,6 ani conform datelor fundației Beyond Batten.
Autismul, epilepsia, ADHD sunt câteva dintre bolile cu care sunt diagnosticați greșit copii cu CLN2.
Noi posibile terapii pentru boli din familia lipofuscinozelor
În august 2017, substanța PLX 200, dezoltată de Polaryx Therapeutics, având ca principală indicație CLN2 a fost desemnată de FDA medicament orfan. Substanța are ca substrat factorul de transcripție EB (TFEB), care în studii pe modele animale a avut rezultate favorabile asupra supraviețurii și funcției motorii.
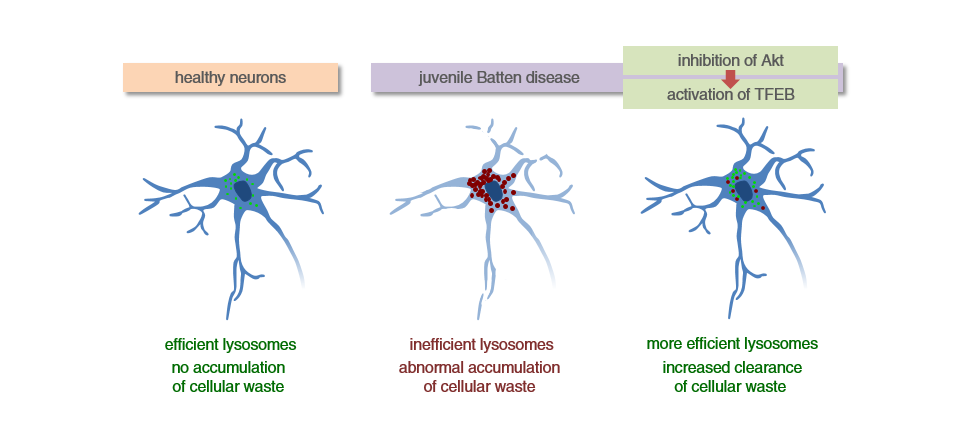
Pentru forma juvenilă, CLN 3 cercetătorii de la Baylor College of Medicine lucrează în prezent la dezvoltarea unui tratament, bazându-se tot pe modularea TFEB. Unul dintre aceștia, Dr. Marco Sardiello, a explicat:
„Cu câțiva ani în urmă s-a identificat o proteină în celule, TFEB, un factor de transcripție care stimulează celula să producă mai mulți lizozomi și astfel să prevină eficient acumularea detritusurilor celulare. Am început astfel să studiem cum să activăm TFEB farmacologic”
Răspunsul a fost trehaloza, un dizaharid care inhibă o altă proteină, Akt, permițând TFEB să determine activarea lizozomală. Prin administrarea aceastei substanțe s-a reușit reducerea inflamației de la nivel cerebral și împiedicarea procesului neurodegenerativ.
Studiul integral poate fi citit – aici
Această descoperire reprezintă un pas important, deoarece identifică un alt mecanism din spatele bolilor de stocaj lizozomal. Mai mult decât atât, încurajează cercetările să se extindă pentru descoperirea tratamentelor eficiente și pentru alte boli care presupun stocarea substanțelor patologice cum ar fi bolile Alzheimer, Parkinson sau Huntington.
National Organization for Rare Disorders (NORD) estimează că pentru aproximativ 50% dintre bolile rare nu există o asociație sau o fundație care să sprijine cercetările pentru găsirea unui tratament. 50% dintre pacienții care suferă de boli rare sunt copii, iar 30% dintre aceștia nu ajung să își sărbătorească aniversarea de 5 ani.
În țara noastră, conform datelor de la Alianţa Naţională pentru Boli Rare din România un milion de oameni suferă de boli rare, dintre care 75% sunt copii, iar 9 din 10 nu sunt diagnosticați sau sunt diagnosticați greșit.