
Cum se acordă autorizarea condiționată de punere pe piață în Uniunea Europeană pentru vaccinurile împotriva COVID-19?
Autorizarea condiționată de punere pe piață (în cazul Agenției Europene a Medicamentului) sau autorizarea în regim de urgență (Emergency Use Authorization, EUA în SUA) pentru vaccinurile contra COVID-19 sunt mecanisme care fac parte din eforturile de combatere a pandemiei și vor determina rapiditatea cu care vaccinul va fi disponibil pe scară largă.
Pentru transparentizarea procesului, agențiile responsabile cu emiterea autorizațiilor în regim de urgență din Statele Unite (Food and Drug Administration, FDA) și din Uniunea Europeană (Agenția Europeană a Medicamentului, EMA) vor susține întâlniri publice pe tema vaccinurilor COVID-19 la începutul lunii decembrie 2020. FDA a programat o reuniune a Comitetului său Consultativ pentru Vaccinuri și Produse Biologice Conexe pe 10 decembrie 2020 pentru a discuta despre solicitarea de emitere a EUA pentru vaccinul COVID-19 produs de Pfizer și BioNTech. Pe 11 decembrie 2020, EMA va organiza o consultare publică pentru a informa cetățenii europeni despre procesele de reglementare ale Uniunii Europene (UE) pentru aprobarea vaccinurilor COVID-19 și rolul agenției în dezvoltarea, evaluarea, aprobarea și monitorizarea siguranței acestora. Ambele întâlniri vor fi difuzate live și le veți putea urmări pe Raportuldegardă.ro.

În cazurile de urgență legate de sănătatea publică precum pandemia SARS-CoV2, cercetarea și dezvoltarea vaccinurilor este accelerată la nivel global, urmând o cale rapidă (fast-track), astfel încât vaccinurile să poată fi puse la dispoziția populației cât mai curând posibil. Dezvoltarea vaccinurilor contra COVID-19 este comprimată în timp, însă este fundamentată pe cunoștințele extinse despre producția de vaccinuri obținute cu vaccinurile pre-existente pandemiei, dar și despre tehnologiile aplicate, care au fost studiate timp îndelungat înaintea aplicării.
Calea de dezvoltare standard a vaccinurilor
Dezvoltarea standard a oricărui vaccin este un proces îndelungat, iar studiile se implementează în următoarele etape secvențiale:
- Compania farmaceutică produce loturi mici și derulează studii la scară mică pentru optimizarea procesului de producție (determinarea formulei adecvate care să poată menține componentele vaccinului stabile până la sfârșitul perioadei de valabilitate);
- Producătorii decid dacă vor continua dezvoltarea vaccinului și extinderea producției sale, dezvoltând totodată o strategie adecvată și eficientă de control al calității. Calitatea farmaceutică este evaluată la fiecare dintre componentele vaccinului, formularea finală care trebuie utilizată și întregul proces de fabricație în detaliu;
- Se efectuează mai multe studii non-clinice, pe modele de laborator, utilizând studii in vitro sau modele pe animale (studii in vivo), pentru a demonstra capacitatea vaccinului de a declanșa un răspuns imun și de a preveni infecția;
- Dezvoltatorul vaccinului studiază produsul în trei faze ale studiilor clinice, înrolând un număr mai mare de voluntari în fiecare fază:
- studiile de fază I (între 20 și 100 de voluntari sănătoși) au scopul de a confirma dacă vaccinul se comportă conform așteptărilor pe baza testelor de laborator pentru a stabili următoarele: dacă vaccinul declanșează răspunsul imun așteptat, dacă vaccinul este sigur să treacă la studii mai ample și care sunt dozele adecvate;
- studiile de fază II (câteva sute de voluntari) investighează cele mai bune doze de utilizat, cele mai frecvente efecte secundare și câte doze sunt necesare. Aceste studii verifică, de asemenea, dacă vaccinul declanșează un răspuns imun optim la o populație mai largă și pot oferi câteva indicații preliminare despre eficacitate;
- studiile de fază III (mii de voluntari) demonstrează rata de eficacitate în protejarea împotriva infecției (comparativ cu placebo sau tratament alternativ) și care sunt efectele secundare mai puțin frecvente la cei care primesc vaccinul în dezvoltare.
- Dacă rezultatele studiilor clinice sunt favorabile, vaccinul demonstrând standarde înalte de eficacitate și siguranță, compania producătoare poate depune toate datele colectate în vederea evaluării de către autoritățile de reglementare din domeniu;
- În cazul obținerii autorizației de comercializare a produsului, compania producătoare va începe procesul de manufacturare al vaccinului aprobat, mărindu-și capacitatea de producție.
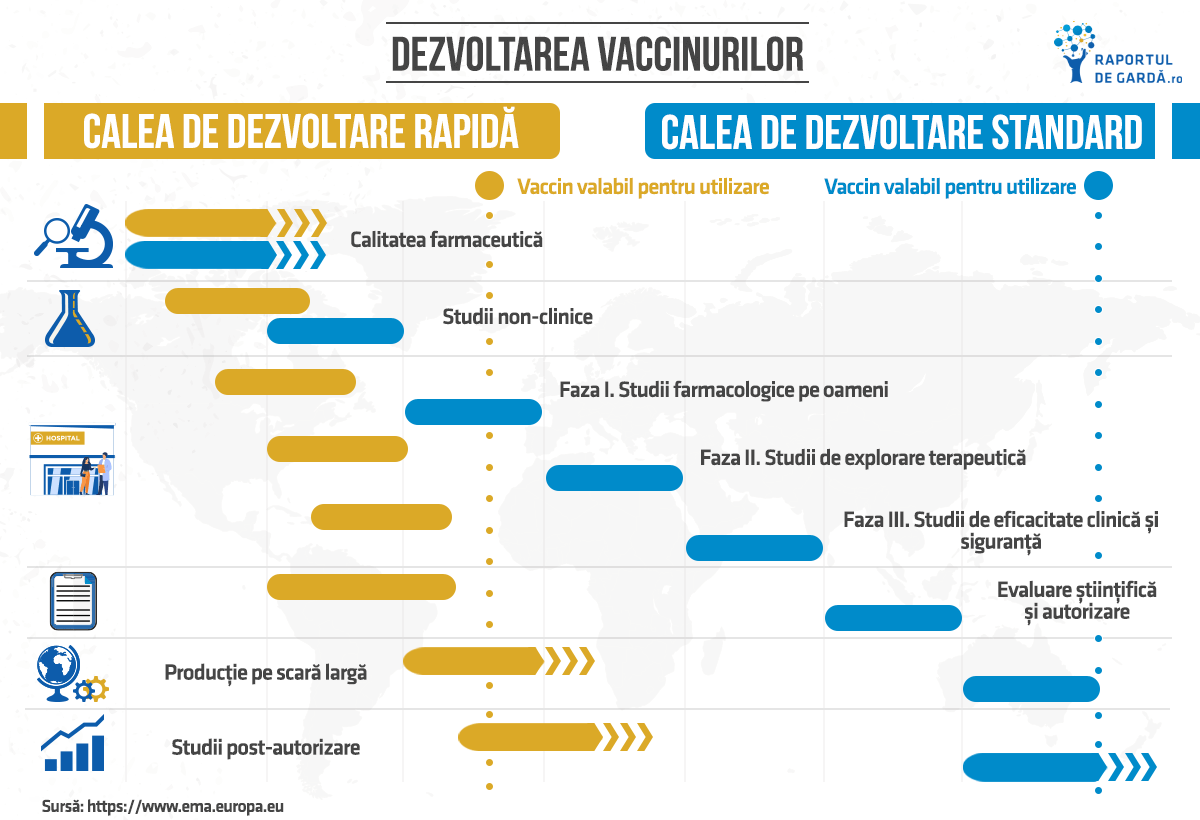
Calea de dezvoltare rapidă a vaccinurilor în caz de urgență de sănătate publică (fast-track)
Ceea ce este diferit pentru vaccinurile COVID-19 este că viteza de dezvoltare și aprobarea potențială sunt mult mai rapide din cauza urgenței de sănătate publică. Spre deosebire de calea standard, diferența majoră constă în faptul că etapele de dezvoltare se suprapun. Acest lucru este posibil deoarece autoritățile de reglementare alocă resurse suplimentare pentru evaluarea pe tot parcursul dezvoltării și cercetării produsului, nu doar la finalizarea acestui proces. Componentele cheie ale dezvoltării vaccinurilor COVID-19 prin calea de dezvoltare rapidă (fast-track) sunt următoarele:
- Standardele de reglementare – vaccinurile COVID-19 trebuie aprobate în conformitate cu aceleași standarde care se aplică tuturor medicamentelor din UE;
- Dezvoltarea – este comprimată în timp, aplicând cunoștințele actuale extinse privind dezvoltarea vaccinului;
- Resursele – dezvoltarea vaccinului COVID-19 mobilizează mai multe resurse simultan;
- Dialogul continuu – dezvoltarea vaccinului COVID-19 este susținută de un dialog timpuriu și continuu între dezvoltatori și un grup dedicat de experți în reglementare;
- Producția – companiile extind capacitatea de producție și manufacturare pentru a asigura disponibilitatea și distribuirea eficientă a vaccinului.
Unele vaccinuri COVID-19 sunt dezvoltate folosind metode noi care se așteaptă să crească volumul și viteza de producție în comparație cu alte tipuri de vaccinuri, să sporească stabilitatea produsului și să producă răspunsuri imune puternice. Alte vaccinuri sunt dezvoltate folosind metodele existente, ceea ce înseamnă că ar putea fi mai ușor să se utilizeze liniile de producție existente pentru a produce vaccinuri COVID-19 la scară largă. Companiile pot utiliza diverse abordări pentru a reduce termenele de dezvoltare, precum:
- mobilizarea simultană a mai multor resurse umane pentru a analiza mai rapid rezultatele studiilor anterioare și pentru a stabili următorii pași în ceea ce privește resursele, finanțarea și strategia de reglementare;
- combinarea fazelor studiilor clinice sau efectuarea unor studii în paralel, acolo unde este sigur.
Companiile extind, de asemenea, capacitatea de producție și producția la scară largă, pentru a facilita disponibilitatea vaccinului fără întârziere după aprobare. În UE, Comisia Europeană a oferit sprijin pentru a facilita dezvoltarea și disponibilitatea vaccinului cât mai repede posibil, prin securizarea unor contracte cu multiple companii farmaceutice dezvoltatoare de potențiale vaccinuri COVID-19.
Evaluarea științifică și aprobarea condiționată
Vaccinurile COVID-19 pot fi aprobate și utilizate numai dacă respectă toate cerințele de calitate, siguranță și eficacitate stabilite în legislația farmaceutică a UE. Având în vedere pandemia, EMA și agențiile de reglementare din Europa redirecționează resurse pentru a accelera procesele și a reduce termenele pentru evaluarea și autorizarea vaccinurilor COVID-19.
Cadrul de reglementare solid și expertiza științifică în UE
Legislația farmaceutică a UE asigură faptul că vaccinurile sunt aprobate numai după ce evaluarea științifică a demonstrat că beneficiile lor generale depășesc riscurile lor. Beneficiile unui vaccin împotriva COVID-19 trebuie să fie mult mai mari decât orice efect secundar sau riscuri potențiale. EMA a creat un grup specializat de experți și proceduri de revizuire rapidă pentru a evalua aplicațiile de înaltă calitate de la companii, în cel mai scurt timp posibil, asigurând în același timp opinii științifice solide. Grupul de lucru COVID-19 (ETF) din cadrul EMA reunește experți cheie din întreaga rețea europeană de reglementare a medicamentelor pentru a asigura un răspuns rapid și coordonat în ceea ce privește pandemia.
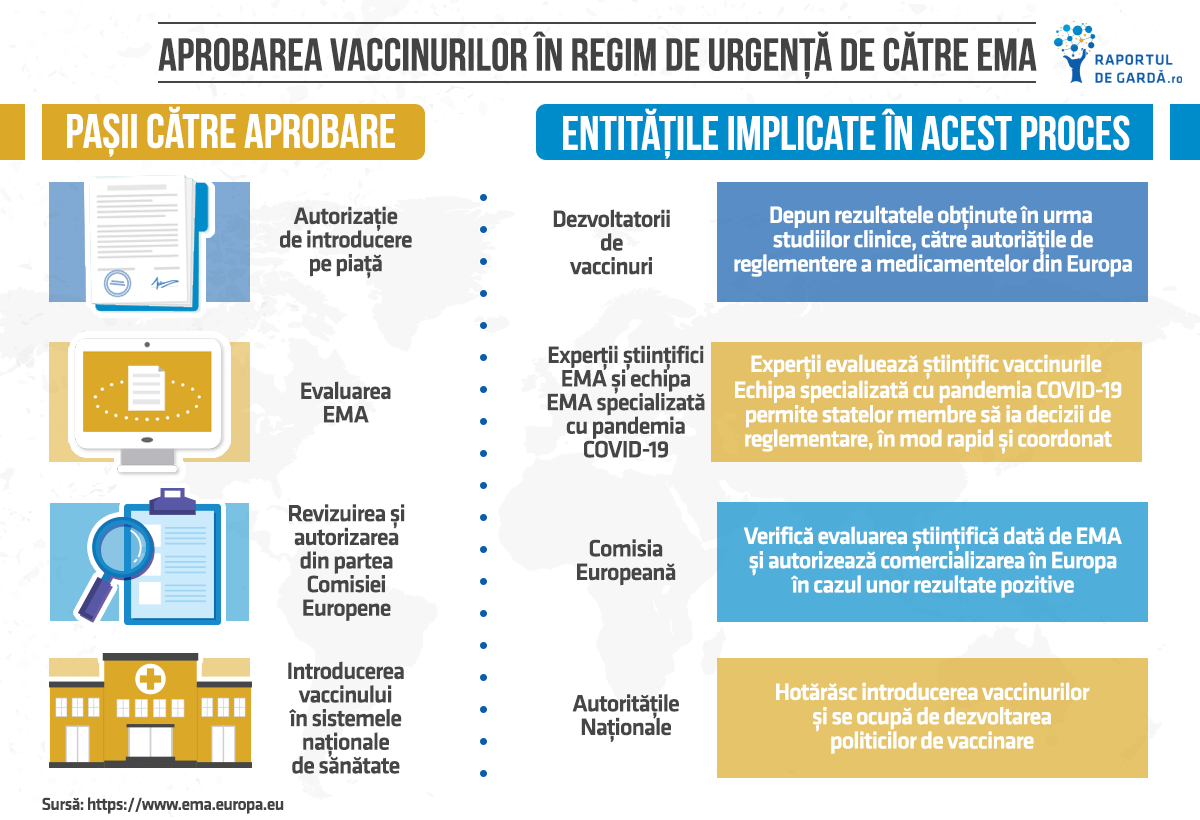
Procesele de evaluare științifică și aprobare
Pentru a obține aprobarea pentru un vaccin în UE, dezvoltatorul transmite rezultatele tuturor studiilor implementate către autoritățile de reglementare a medicamentelor din Europa. Aceasta face parte dintr-o cerere de autorizație de introducere pe piață, iar în cazul candidaților pentru vaccin COVID-19, trebuie să ofere detalii despre:
- grupul de persoane cărora li se va administra vaccinul;
- calitatea farmaceutică a acesteia, inclusiv informații privind identitatea și puritatea componentelor vaccinului și conținutul și activitatea biologică (potență);
- date privind fiecare etapă de fabricație și controalele utilizate pentru a se asigura că fiecare lot de vaccin este în mod constant de bună calitate;
- respectarea cerințelor internaționale pentru testarea în laborator, fabricarea vaccinurilor și desfășurarea studiilor clinice;
- tipuri de răspunsuri imune induse de vaccin;
- efectele observate în grupurile de persoane cărora li se va administra vaccinul;
- efectele secundare ale vaccinului, observate la cei vaccinați, inclusiv dacă există date în rândul populațiilor speciale, cum ar fi vârstnicii sau femeile însărcinate;
- informații destinate a fi colectate din studii ulterioare după autorizare (de exemplu, date de siguranță pe termen lung sau imunitate pe termen lung);
- prescrierea informațiilor care trebuie furnizate pacienților și profesioniștilor din domeniul sănătății (adică rezumatul caracteristicilor produsului, etichetarea și prospectul), care sunt elaborate de dezvoltator și revizuite și agreate de comitetele științifice ale EMA;
- modul în care riscurile vor fi gestionate și monitorizate odată ce vaccinul este autorizat;
- planul de gestionare a riscurilor (risk management plan), un document cu informații despre orice posibile (cunoscute sau potențiale) probleme de siguranță ale vaccinului, modul în care riscurile vor fi gestionate și monitorizate odată ce vaccinul este autorizat și ce informații se intenționează a fi colectate din următoarele studii.
Majoritatea vaccinurilor COVID-19 din UE vor fi evaluate de EMA prin procedura centralizată (care este obligatorie pentru orice vaccin care utilizează biotehnologie și presupune o singură aplicație, o singură evaluare și o singură autorizație pentru întreg teritoriul UE).
Evaluarea accelerată
Conform legislației farmaceutice a UE, calendarul standard pentru evaluarea unui medicament este de maximum 210 zile active. Cu toate acestea, pentru vaccinurile COVID-19, EMA aplică o procedură accelerată, numită revizuire continuă (rolling review).
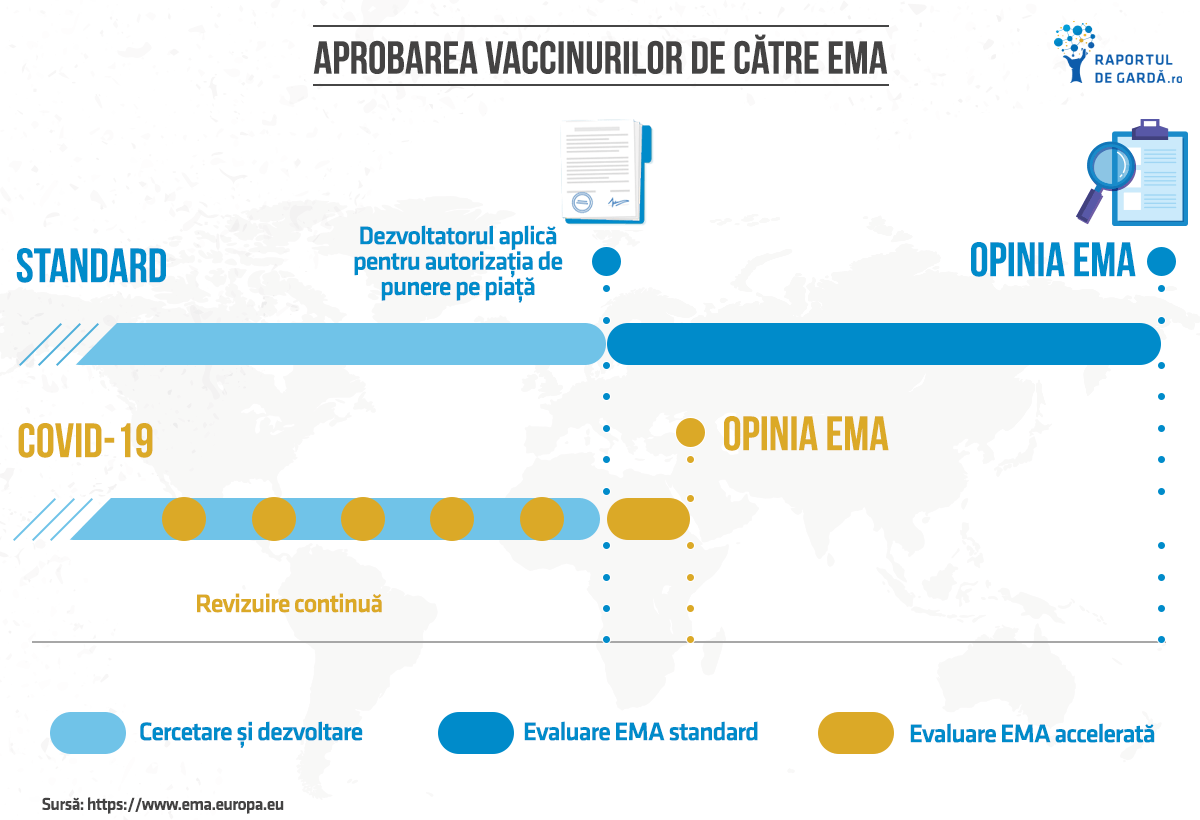
Acest lucru permite EMA să înceapă evaluarea datelor pentru un potențial vaccin promițător imediat ce acestea devin disponibile. Când evaluarea este finalizată, EMA are opțiunea de a recomanda o autorizație condiționată de introducere pe piață, un tip de aprobare pentru vaccinurile care răspund nevoilor medicale nesatisfăcute și, în special, pentru a fi utilizate în situații de urgență ca răspuns la amenințările pentru sănătatea publică recunoscute de Organizația Mondială a Sănătății (OMS) sau de către UE. Rezumatul caracteristicilor produsului (prospectul), care este redactat de către agenția de reglementare pe baza datelor furnizate de către producători, va fi publicat și diseminat după ce vaccinul COVID-19 primește aprobarea. Prospectul va avea la bază analiza datelor disponibile referitoare la calitatea procesului de manufacturare, siguranța și eficacitatea vaccinului COVID-19.
EMA a primit deja cererea de autorizare condiționată de introducere pe piață pentru BNT162b2, vaccinul COVID-19 dezvoltat de BioNTech și Pfizer. Autoritățile de reglementare Europene au anunțat comunicarea concluziilor evaluării vaccinului până la cel târziu 29 decembrie 2020, dat fiind revizuirea continuă ce a avut loc până în prezent.
Comisia Europeană va evalua opiniile științifice ale grupului de experți EMA și, în cazul unor concluzii favorabile, va emite o autorizație de utilizare pe întreg teritoriul UE. Ulterior, autoritățile locale ale fiecărui stat membru vor decide asupra politicilor și strategiilor de vaccinare naționale.
Monitorizarea siguranței și utilizării vaccinului post-autorizare
Ca orice medicament, vaccinurile prezintă beneficii și riscuri. Niciun vaccin nu este sută la sută eficient în prevenirea bolilor sau sută la sută sigur la toate persoanele vaccinate. Anumite reacții adverse rare sau foarte rare pot apărea numai atunci când milioane de oameni sunt vaccinați. După aprobarea unui potențial vaccin COVID-19, un număr foarte mare de persoane vor primi vaccinul, iar legislația UE impune monitorizarea siguranței vaccinurilor în timpul utilizării acestora.
Siguranța vaccinurilor COVID-19 va fi monitorizată conform ghidului uzual de bune practici în farmacovigilență (monitorizarea standard), dar și prin activități suplimentare specifice pentru vaccinările anti SARS-CoV2 (vezi planul de monitorizare a siguranței și ghidul de management al riscurilor pentru vaccinurile contra COVID-19). În contextul pandemiei, autoritățile de reglementare și dezvoltatorii de vaccinuri mobilizează resurse suplimentare pentru a monitoriza siguranța și a gestiona riscurile vaccinurilor contra COVID-19. În prezent, deoarece virusul este atât de nou, nu există suficiente cunoștințe despre cât va dura imunitatea conferită după vaccinare sau dacă va fi nevoie de doze de rapel periodice. Datele din studiile de imunogenitate și eficacitate pe termen lung vor informa viitoarele strategii de vaccinare.
Situații anterioare în care a fost utilizată autorizarea condiționată de punere pe piață
Autorizarea condiționată de punere pe piață reprezintă un instrument important pentru autoritățile din domeniul sănătății publice pentru cazurile de urgență, deoarece le poate permite să utilizeze cea mai bună măsură de control disponibilă pentru a detecta, preveni sau trata o boală care se răspândește rapid în comunitate.
Mecanismele pentru reglementarea obținerii unei autorizații în regim de urgență nu au fost create odată cu apariția virusului SARS-CoV2 sau declararea pandemiei COVID-19. Aceste proceduri erau deja prevăzute în modalitățile de operare ale autorităților din domeniu (atât EMA, cât și FDA) și au fost utilizate în trecut pentru aprobarea de urgență a mai multor produse farmaceutice.
În Statele Unite, programul EUA a fost înființat în 2004, când Proiectul BioShield Act, printre alte măsuri, a modificat și secțiunea 564 din Legea federală privind alimentele, medicamentele și produsele cosmetice pentru a include procedurile de autorizație în regim de urgență. Printre primele EUA emise de către FDA s-au numărat cele venite ca răspuns în fața pandemiei cu H1N1 din anul 2009. Multiple alte produse farmaceutice au fost autorizate printr-o cale rapidă de evaluare și în UE, un exemplu fiind crizotinib, medicament aprobat în 2012 de către EMA pentru tratamentul unor forme de cancer pulmonar. Crizotinib a primit autorizația de comercializare condiționată la numai aproximativ 18 luni de la începutul studiilor de fază I și este utilizat și în prezent.
În data de 2 decembrie 2020, prima autorizație pentru utilizare în regim de urgență acordată pentru un vaccin împotriva COVID-19 a fost emisă de către autoritățile din Marea Britanie. Campania de imunizare contra COVID-19 a început pe 8 decembrie 2020 în rândul populației din Marea Britanie. Guvernul britanic a publicat și prospectul pentru vaccinul COVID-19 produs de Pfizer/BioNTech pe care l-au aprobat.
Citește și:
- Esențial COVID-19: cele mai importante știri ale săptămânii. Costurile nevăzute ale măsurilor de lockdown
- Barometrul de Sănătate Publică Octombrie 2020: Ce cred românii despre vaccinarea împotriva COVID-19?
- EMA: planul de monitorizare a siguranței și ghidul de management al riscurilor pentru vaccinurile COVID-19