
PREMIERĂ. Primul copil din UK a primit terapia genică pentru leucodistrofie metacromatică în afara studiilor clinice
NHS England a anunțat pe 15 februarie că primul copil din UK cu leucodistrofie metacromatică, o boală metabolică rară, a fost tratat în afara studiilor clinice cu terapia genică atidarsagene autotemcel (denumire comercială – Libmeldy) și are o evoluție corespunzătoare vârstei sale. Viața lui Teddi, o fetiță în vârstă de 19 luni acum, a fost astfel salvată și se așteaptă că va continua la fel ca a unui copil sănătos. La vârsta de un an, celulele stem ale fetiței au fost prelevate, modificate genetic, iar în august 2022 au fost reintroduse în organismul acesteia. Fetița a fost externată în octombrie, iar în momentul actual, simptomele bolii au dispărut, conform părinților.
Traseul către aprobarea terapiei genice a început încă din 2007, când a primit statutul de medicament orfan în Uniunea Europeană, culminând în octombrie 2020 cu recomandarea CHMP pentru aprobarea punerii pe piață, și, ulterior, aprobarea în sine, în decembrie același an.

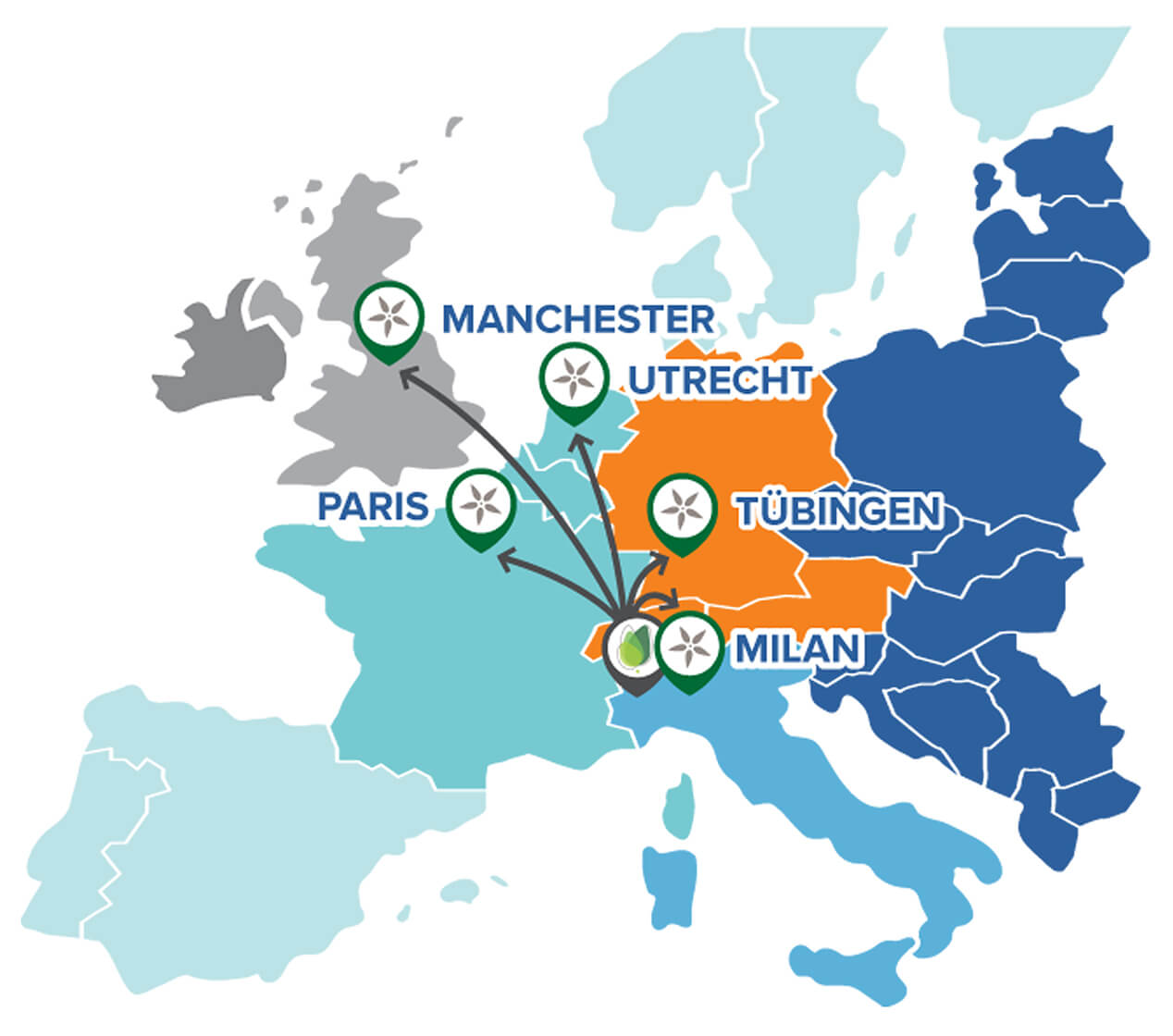
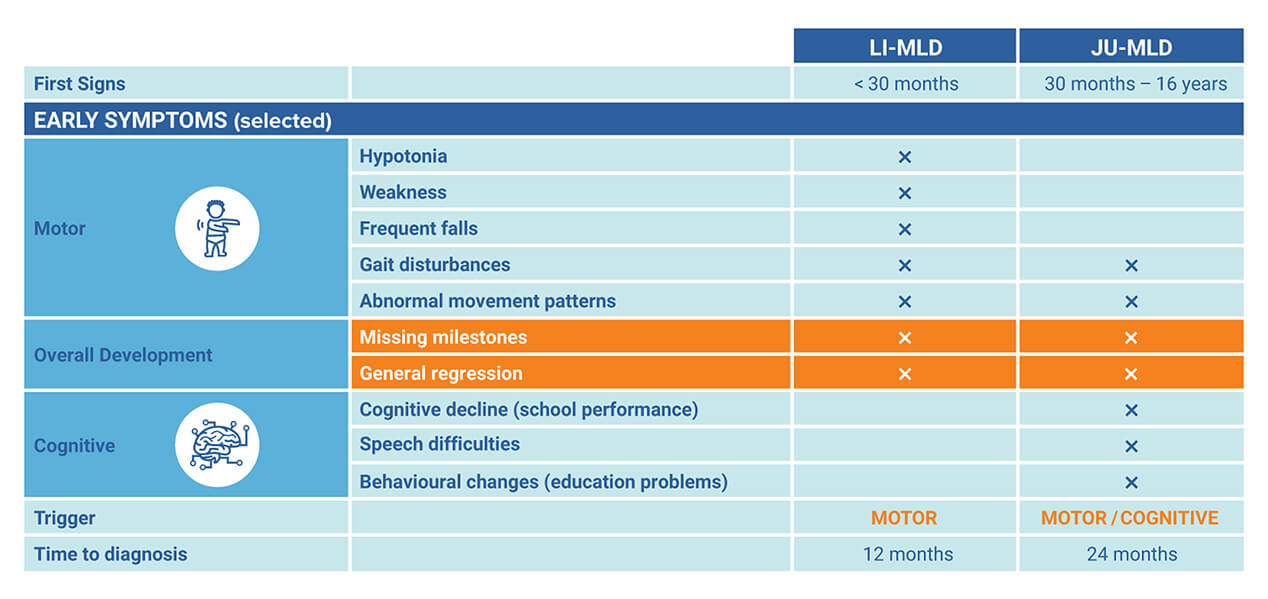
Tratamentul este disponibil în UK prin NHS (National Health Service) ca serviciu de specialitate și este livrat în cadrul Spitalului de Copii Royal Manchester – în colaborare cu Centrul de Medicină Genomică din Manchester – ambele făcând parte din Manchester University NHS Foundation Trust. Centrul din Manchester este unul dintre cele cinci site-uri europene care administrează tratamentul și singurul loc din Marea Britanie. Aproximativ cinci copii se nasc cu MLD în fiecare an în Anglia. Cei a căror boală debutează înainte de 30 de luni se deteriorează rapid și de obicei mor între cinci și opt ani. Cei cu debut tardiv – semnele bolii apar între 30 de luni și șase ani – au o speranță de viață cu 10 până la 20 de ani mai mult.
Leucodistrofia metacromatică (deficienţa de arilsulfataza A, abreviată în engleză ca MLD) este o boală metabolică rară care afectează sistemul nervos și determină pierderea progresivă a funcțiilor motorii și cognitive. Boala este cauzată de mutații bialelice la nivelul genei ARSA, ceea ce conduce la o scădere a activității enzimei codificate de această genă, iar consecința este acumularea de sulfatide la nivel cerebral și în alte zone ale organismului (ficat, vezică biliară, rinichi, splină), precum și distrugerea mielinei de la nivelul sistemului nervos central și periferic. Boala este împărțită în 3 forme, în funcție de vârsta la care debutează simptomele (infantilă tardivă, juvenilă și o formă care apare la adulți).
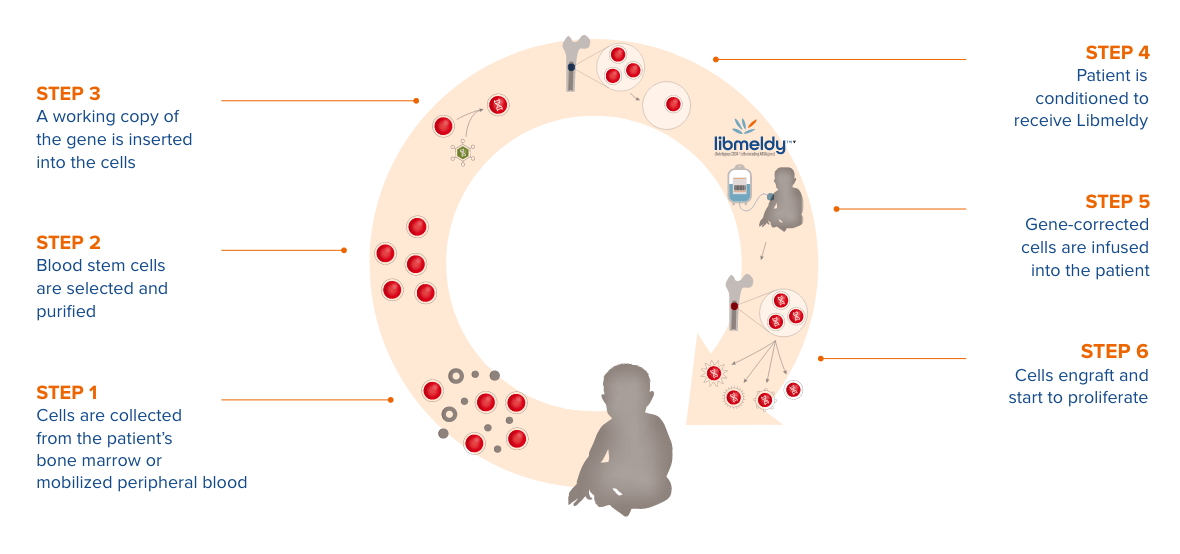
Libmeldy este o terapie genică ex vivo, care implică prelevarea și modificarea celulelor stem hematopoietice ale pacientului. Cu ajutorul unui vector viral sunt inserate copii funcționale ale genei ARSA la nivelul genomului celulelor pacientului, care sunt ulterior reintroduse în organism. Studiul care a stat la baza aprobării în Uniunea Europeană a urmărit 35 de pacienți, pe o perioadă de până la 8 ani, iar rezultatele au arătat eficacitatea în timp a terapiei.
O singură administrare intravenoasă a Libmeldy a modificat semnificativ evoluția bolii pentru majoritatea pacienților cu leucodistrofie metacromatică în formele cu debut pediatric (infantil tardiv și juvenil precoce).
De ce screeningul nou-născuților și diagnosticarea precoce pot face diferența dintre viață și moarte
Teddi nu este singura din familie diagnosticată cu MLD. Sora sa mai mare, Nala, care acum are 3 ani, a fost de asemenea diagnosticată în aprilie 2022, în același timp cu Teddi, dar boala progresase prea mult pentru a mai fi eligibilă pentru terapie.
Câteva declarații de la mama celor două surori, din comunicatul oficial NHS: „În aprilie anul trecut, lumea noastră a fost dată peste cap când ambele noastre fetițe au fost diagnosticate cu MLD. Să ni se spună că prima noastră fiică, Nala, nu era eligibilă pentru niciun tratament, că va continua să-și piardă toate funcțiile și că va muri extrem de tânără a fost cel mai sfâșietor și mai greu de acceptat. Cu toate acestea, aveam speranțe pentru fiica noastră mai mică, Teddi. Ni s-a spus că, din fericire, un nou tratament de tip terapie genică a fost pus la dispoziție recent prin NHS. Suntem extrem de privilegiați că Teddi este primul copil din UK care a primit această terapie și recunoscători că are oportunitatea de a duce o viață lungă și, sperăm, normală. Fără acest tratament, ne-am confrunta cu pierderea ambilor noștri copii.
Putem doar să sperăm că într-o zi, un tratament va deveni disponibil pentru toate etapele MLD și susținem cu tărie că ar trebui ca nou-născuții să fie testați pentru a salva mai multe familii de la a trece prin această durere”.
În forma infantilă tardivă, mortalitatea la 5 ani de la debut se estimează a fi la 50%. MLD este moștenită printr-un model autozomal-recesiv – frecvența purtătorilor în populația generală fiind între 1:100 și 1:130. Simptomele, vârsta la debut și evoluția bolii variază, dar pacienții progresează spre disfagie (dificultate în înghițirea alimentelor), dizabilitate neurologică severă și deces. Simptomele precoce sunt nespecifice și adesea dificil de identificat, ceea ce duce la trimiteri întârziate către centrele de specialitate.
Întârzierea dezvoltării neurologice în multe alte boli neurodegenerative poate imita MLD – de exemplu, deficitul de Saposina B, boala Krabbe și sindromul Angelman. Ideal este identificarea pacienților pre-simptomatici și simptomatici precoce, ceea ce face screeningul nou-născuților esențial în boli ca MLD.
Procesul de diagnosticare începe cu semnele și simptomele prezentate mai jos, iar pentru confirmarea diagnosticului se utilizează testări biochimice, precum teste privind activitatea enzimei ASRA și teste de urină, și genetice, prin paneluri specifice sau multi-genice. În formele deja avansate sunt utilizate și metodele imagistice.
În decembrie 2022, Guvernul UK anunța investiții de 175 de milioane de lire sterline pentru cercetarea genomică inovatoare pentru a diagnostica bolile rare și cancerul mai rapid și mai precis, dar și pentru a reduce inegalitățile în materie de sănătate și pentru a îmbunătăți înțelegerea ADN-ului și a impactului acestuia asupra sănătății populației. Citiți toate detaliile: Guvernul UK, investiții record pentru screeningul nou-născuților prin secvențierea întregului genom
În cadrul ediției din 21 februarie 2023 a emisiunii Știința360, Radio România Cultural, Dr. Marius Geantă, președinte Centrul pentru Inovație în Medicină, a discutat despre această premieră a lumii medicale:
Citește și:
- Parteneriat BioNTech – UK: 10 mii de pacienți cu cancer ar putea beneficia de imunoterapii personalizate ARNm până în 2030
- Declarația de la Praga: e momentul ca toate țările UE să implementeze programe de screening pediatric pentru hipercolesterolemia familială