
Prima terapie genică pentru pacienții cu beta-talasemie, aproape de aprobare în Uniunea Europeană
Zynteglo este o terapie genică destinată pacienților cu forme grave de beta talasemie, care impun dependența de transfuzii de sânge pe termen lung. Această nouă terapie se administrează o singură dată în viață și presupune introducerea unor copii funcționale ale genei de beta globină în celulele stem hematopoietice recoltate de la pacient.
Comitetul pentru Produse Medicamentoase de Uz Uman (CHMP) din cadrul Agenției Europene a Medicamentului recomandă acordarea autorizației de punere pe piață condiționată, iar în următoarele luni se așteaptă decizia oficială din partea Comisiei Europene.

„Scopul tratamentului cu Zynteglo este să permită pacienților cu beta talasemie să aibă o producție normală de hemoglobină, astfel încât să obțină independența față de transfuziile sanguine repetate” – David Davidson, Directorul Departamentului Medical de la Bluebird, compania care produce Zynteglo
Rezultatele studiilor pe care se bazează cererea de aprobare
Tratamentul este indicat pentru pacienții cu beta talasemie, dependenți de transfuzie, cu vârste de peste 12 ani, care nu prezintă un genotip β0/β0 și sunt eligibili pentru un transplant de celule stem hematopoietice, dar pentru care nu există disponibil un donator compatibil. Recomandarea se bazează pe datele mai multor studii de fază 1/2 și de fază 3, unele dintre ele fiind prezentate anul trecut la Congresul Societății Americane de Hematologie (ASH 2018).
Studiului de fază 1/2, Northstar a arătat că pentru pacienții la care s-a administrat terapia genică, după o perioadă de urmărire de peste 3 ani, nu mai sunt necesare transfuziile regulate de sânge pe care pacienții le primeau anterior iar hemoglobina se menține la valori stabile.
- Independența față de transfuzii s-a înregistrat la 8 din 10 pacienți fără genotip β0/β0 (independența s-a definit prin lipsa necesității de transfuzie pentru cel puțin 12 luni, iar hemoglobina s-a menținut la valori de peste 9 g/dl)
Studiul de fază 3, Northstar-2, este încă în derulare, însă au fost aduse îmbunătățiri în procesul de obținere a terapiei. Primele date arată că pacienții pot atinge valori aproape normale ale hemoglobinei fără necesitatea transfuziilor.
- La 6 luni după administrarea ZYNTEGLO, valoarea medie a hemoglobinei determinată la grupul de pacienți a fost de 11,9 g/dl ( conform datelor prezentate în septembrie 2018)
Studiul de fază 3, Northstar 3 a evaluat pacienți cu forme mai grave de boală, unde obiectivele au fost concentrate pe reducerea numărului de transfuzii. Conform datelor din noiembrie 2018, cei trei pacienți evaluați la acel moment prezentau valori medii ale hemoglobinei de peste 10 g/dl.
Terapia se adresează și pacienților cu anemie falciformă, pentru care este evaluată într-un studiu de fază 1/2.
De ce este importantă terapia genică pentru controlul bolilor hematolgice rare?
Beta talasemia este o boală genetică ce presupune anomalii ale unei subunități a hemoglobinei, proteina din hematii responsabilă de transportul oxigenului la țesuturi. Beta talasemia și anemia falciformă (siclemia) sunt cele mai frecvente boli hematologice ereditare, ambele fiind caracterizate de mutații la nivelul genei globinei. Siclemia presupune o singură mutație a acestei gene, care conduce la apariția hemoglobinei S. Beta talasemia implică una sau mai multe dintre cele peste 300 de mutați în gena beta globinei. În funcție de aceste mutații pot apărea fenotipuri asociate cu forme mai ușoare de boală, în care pacienții nu depind de transfuzii și forme severe, cum e beta talasemia majoră.
Pacienții cu o formă severă a bolii prezintă supraîncărcare cu fier, anomalii scheletice, complicații cardiace, disfuncție hepatică și afectare endocrină.
Pacienții cu beta talasemie majoră au un prognostic mai rezervat și necesită monitorizare continuă. Aceștia primesc regulat transfuzii de sânge pentru controlul anemiei, însă transfuziile cronice determină supraîncărcarea cu fier, care are un impact negativ asupra organelor vitale. Dozarea fierului și administrarea chelatorilor de fier, medicamente care facilitează eliminarea acestui compus, sunt măsuri uzuale pentru pacienți.
„Pentru pacienții mei, beta talasemia înseamnă o viață care depinde de transfuzii de sânge, terapia cu substanțe chelatoare de fier, controlul anemiei și al complicațiilor. Povara asupra pacienților și a familiilor lor este semnificativă, astfel încât spitalizările, asistența medicală pe termen lung și simptomele au un impact major asupra calității vieții” – Prof. Franco Locatelli, Universitatea Sapienza, Roma
Singura variantă terapeutică cu potențial curativ pentru beta talasemie este transplantul allogen de celule stem hematopoietice. Transplantul de la un donator înrudit, compatibil cu pacientul determină supraviețuiri de peste 80% și se asociază cu mortalitate de 3-10% pe termen lung, însă se găsesc donatori în mai puțin de 30% dintre cazuri. Transplantul de la donatori compatibili, dar care nu sunt înrudiți cu pacientul cresc mortalitatea și scad supraviețuirea. În schimb, aproape toți pacienții ar obține beneficii prin terapia genică deoarece propriile celule ar fi programate să producă o formă funcțională a hemoglobinei iar toxicitatea ar fi eliminată.
Etape pentru accesul la terapia genică pentru beta talasemie
Bluebird se pregătește de lansarea terapiei în Europa, ceea ce presupune o serie de măsuri speciale și crearea cadrului instituțional potrivit. În primul rând, sunt necesare centre specializate în colectarea celulelor stem hematopoietice de la pacienți, printr-un proces numit afereză. Prelucrarea celulelor trebuie realizată în condiții optime de laborator. Un vector viral va fi folosit pentru introducerea genei modificate pentru globină β A-T87Q în celulele stem hematopoietice ale subiectului, etapă numită transducție. Înainte de infuzia celulelor stem modificate înapoi la pacient, măduva hematogenă este pregătită prin intermediul chimioterapiei.
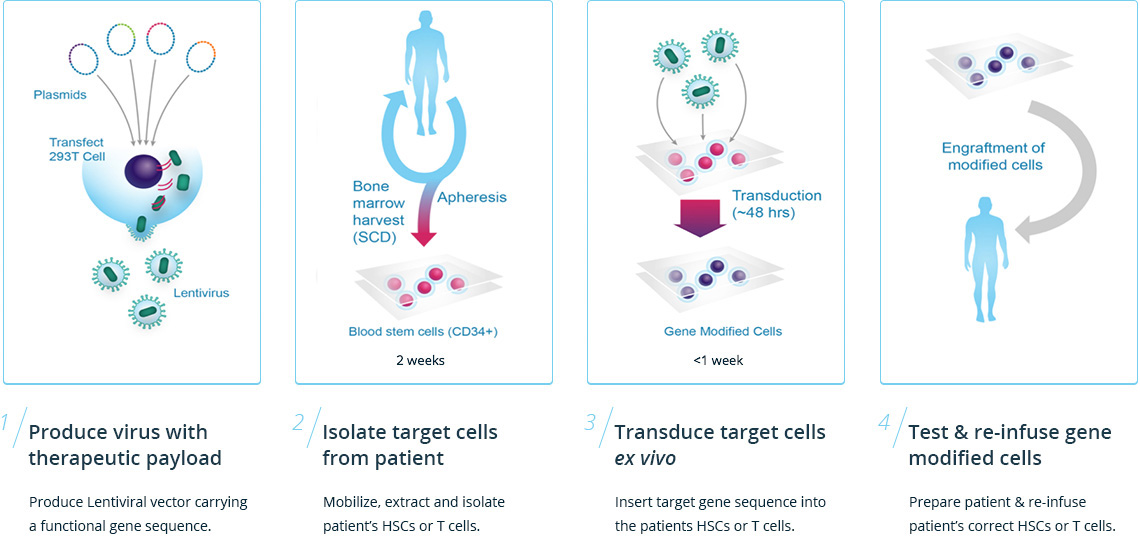
Compania a depus cererile de aprobare mai întâi în Europa și nu în Statele Unite având în vedere datele legate de incidența bolii, dar și luând în considerare mecanismele accelerate pe care EMA le folosește tot mai frecvent pentru a facilita accesul la terapii inovatoare. Beta talasemia este mai frecventă în zona Mediteraneană, Orientul Mijlociu, Africa și Asia și mai rar întâlnită în Statele Unite. În 2020 este de așteptat ca terapia să ajungă și în atenția Food and Drug Administration.
„Programele inovative dezvoltate de Agenția Europeană a Medicamentului sunt esențiale pentru asigurarea accesului precoce la noile terapii pentru pacienții cu afecțiuni pentru care soluțiile sunt limitate” – Anne-Virginie Eggimann, reprezentant Bluebird.
Citește și:
- ISTORIC. 5 terapii genice, rezultate încurajatoare publicate în ultimele 2 luni
- BREAKING NEWS. Luxturna, prima terapie genică aprobată de FDA pentru o boală ereditară care determină orbire