
FDA aprobă selumetinib, prima terapie țintită pentru tratamentul copiilor cu neurofibromatoză tip 1, o afecțiune neurologică rară
FDA (Food and Drug Administration) a aprobat selumetinib pentru tratamentul pacienților cu vârsta de peste 2 ani cu neurofibromatoză de tip 1 (NF1), cu neurofibroame plexiforme simptomatice, inoperabile. Acesta este primul medicament aprobat pentru a trata această afecțiune neurologică rară, întrucât tratamentele anterioare se adresau numai gestionării simptomelor. Koselugo (selumetinib) reprezintă un progres major în tratamentul pacienților cu NF1, deoarece a demonstrat că poate micșora tumorile și poate crește calitatea vieții copiilor cu această boală debilitantă.
„Viața de zi cu zi a tuturor a fost perturbată în timpul pandemiei COVID-19 și, în acest moment critic, dorim ca pacienții să știe că rămânem dedicați și continuăm să considerăm o prioritate maximă pacienții cu tumori rare și boli care pun viața în pericol, precum și nevoile lor unice. Continuăm să accelerăm dezvoltarea produselor medicale pentru acești pacienți”, a declarat Dr. Richard Pazdur, director al Centrului de Excelență Oncologic al FDA, director interimar al Biroului de Boli Oncologice din Centrul pentru Evaluarea și Cercetarea Medicamentelor al FDA.
Studiul clinic SPRINT Stratum 1
Aprobarea FDA s-a bazat pe rezultatele studiului clinic de fază 2 SPRINT Stratum 1, care au fost publicate în revista științifică The New England Journal of Medicine. Pacienții studiați au fost împărțiți în 2 subgrupuri:

- Stratum 1 – pacienți cu cel puțin o complicație legată de neurofibrom;
- Stratum 2 – pacienți fără complicații semnificative din punct de vedere clinic, dar cu potențial de apariție a unei complicații – a căror rezultate nu au fost incluse în studiul de față.
În sugrupul Stratum 1, 50 de pacienți cu NF1 (vârsta medie – 10 ani), au primit selumetinib sub formă de monoterapie orală, 25 mg/m2 de două ori pe zi, în cicluri de câte 28 de zile. Tratamentul a fost continuat timp de 2 ani; iar pacienții care au înregistrat un răspuns parțial sau au avut boală progresivă la momentul de începere a studiului au putut continua tratamentul și după 2 ani.
În acest grup, 66% (33) dintre pacienți au înregistrat răspuns parțial – reducerea cu cel puțin 20% a volumului tumorii. La 82% dintre aceștia răspunsul a durat 12 luni sau mai mult. Cu toate acestea, nu s-au obținut răspunsuri complete în cazul niciunui pacient.
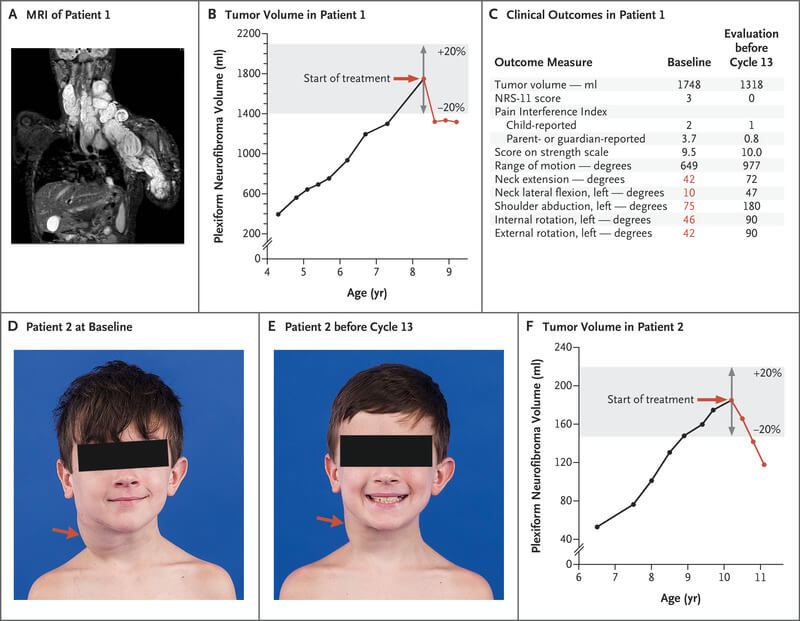
Alte rezultate clinice obținute de pacienți în timpul tratamentului cu Koselugo au constat în ameliorarea leziunilor cutanate desfigurante produse de neurofibroamele plexiforme, a simptomelor și deficitelor funcționale. Deși dimensiunile eșantionului de pacienți evaluați pentru fiecare morbiditate cauzată de neurofibroamele plexiforme (precum desfigurare, durere, rezistență și probleme de mobilitate, compresia căilor respiratorii, deficite de vedere și disfuncția vezicii urinare sau difuncția intestinală) au fost mici, a părut să existe o tendință de îmbunătățire a simptomelor și deficitelor funcționale legate de neurofibroamele plexiforme în timpul tratamentului.
Cele mai frecvente reacții adverse raportate au fost: erupții cutanate, vărsături, dureri abdominale, diaree, oboseală, dureri musculo-scheletale, febră, cefalee. Întreruperea administrării dozelor, reducerea dozelor, și chiar întreruperea permanentă a tratamentului au fost necesare în cazul a 80%, 24% și, respectiv, 12% dintre pacienți. Reacțiile adverse severe au inclus cardiomiopatie, toxicitate oculară, toxicitate gastrointestinală, creștere a creatin-fosfokinazei, niveluri crescute de vitamina E și risc de sângerare.
„În trecut nu existau medicamente aprobate pentru această afecțiune medicală. Aprobarea aceasta are potențialul de a schimba modul în care sunt tratate neurofibroamele plexiforme simptomatice și inoperabile; oferă noi speranțe pacienților cu neurofibromatoză tip 1″, a declarat Prof. Dr. Roy Baynes, vicepreședinte, șef al departamentului de dezvoltare clinică globală al Laboratoarelor de Cercetare Merck.
Managementul dificil al neurofibromatozei tip 1
NF1 este o afecțiune genetică rară și debilitantă, care afectează una din fiecare 3.000 – 4.000 de persoane. Afecțiunea este cauzată de o mutație spontană sau moștenită la nivelul genei NF1 care codifică neurofibromina – o proteină exprimată în special în celulele neuronale. Neurofibromina inhibă calea RAS, ce reglează proliferarea și diferențierea celulelor.
Semnele sugestive pentru diagnostic depind de vârsta la care se manifestă. Riscul de complicații severe din primii ani de viață transformă diagnosticul precoce într-un element crucial, dar și unul dintre motivele pentru care managementul copiilor cu NF1 este dificil.
Neurofibromatoza tip 1 se asociază cu numeroase simptome, inclusiv tumori moi la și sub suprafața pielii (neurofibromatoză cutanată), pigmentarea pielii (așa-numitele „pete de cafea cu lapte”), iar, la 30-50% dintre pacienți, tumorile se dezvoltă pe tecile nervoase (neurofibroame plexiforme). Aceste neurofibrome plexiforme pot provoca probleme clinice, cum ar fi deformări, disfuncții motorii, dureri, disfuncții ale căilor respiratorii, deficiențe de vedere și disfuncții ale vezicii urinare / intestinului. Neurofibroamele plexiforme debutează în timpul copilăriei timpurii, cu diferite grade de severitate și pot reduce speranța de viață cu până la 15 ani.
Neurofibromatoza nu poate fi vindecată, dar tratamentele pot ajuta la gestionarea semnelor și simptomelor (în cazul tumorilor – chirurgie și/sau radioterapie, managementul durerii, chimioterapie în cazul glioamelor optice, managementul tulburărilor psihiatrice, etc.). În general, cu cât copilul se află mai devreme sub îngrijirea unui medic instruit în tratarea neurofibromatozei, cu atât evoluția este mai favorabilă. Tratamentul pentru neurofibromatoza tip 1 (NF1) presupune monitorizare periodică și poate include fizioterapie, sprijin psihologic și managementul durerii. Orice problemă este tratată de o echipă de medici specialiști. Majorității copiilor cu NF1 li se recomandă să efectueze un examen medical complet în fiecare an.
Neurofibroamele pot să nu necesite niciun tratament dacă sunt mici. Cu toate acestea, tratamentul este necesar dacă neurofibromele sunt dureroase sau au impact emoțional asupra copilului. În funcție de mărimea și localizarea neurofibromelor, acestea pot fi tratate cu chirurgie laser sau prin electro-cauterizare. În cazul unor neurofibroame mari, este necesară îndepărtarea chirurgicală a tumorii și chirurgia plastică.
Chirurgia neurofibroamelor plexiforme poate fi mai dificilă. Acest lucru se datorează faptului că aceste tipuri de tumori se răspândesc adesea în țesutul din apropiere și pot afecta structuri osoase importante. Uneori, postoperator, poate apărea deteriorarea nervilor, ceea ce duce la complicații precum pierderea sensibilității sau incapacitatea de a mișca o parte a corpului.
Până acum nu au fost disponibile tratamente standardizate specifice pentru NF1 și complicațiile sale. În ultimii ani, pe măsură ce mecanismele fiziopatogenetice ale bolii au fost înțelese și cunoscute mai bine, a fost posibilă căutarea științifică a unor agenți biologici vizați pentru a schimba cursul bolii.
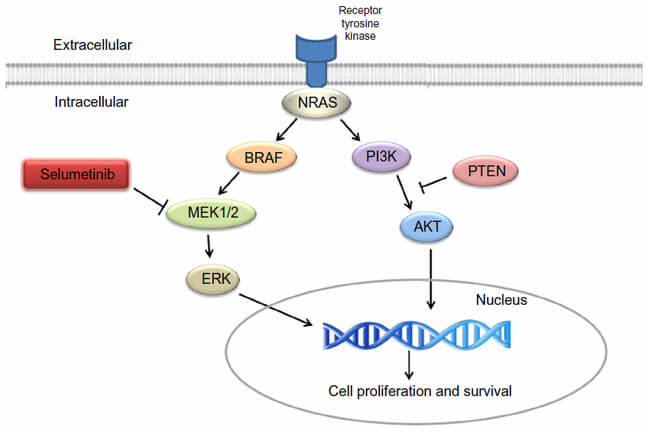
Koselugo (selumetinib) este un inhibitor al kinazei, ceea ce înseamnă că funcționează prin blocarea unei enzime-cheie, și contribuie astfel la inhibarea dezvoltării celulelor tumorale. Inhibă protein-kinazele activate mitogen 1 și 2 (MEK1/2), care sunt reglatori în amonte ale kinazei ERK (extracellular signal-related kinase). Atât MEK cât și ERK sunt componente vitale ale căii RAF-MEK-ERK reglată de RAS – care este adesea activată în diferite tipuri de cancer.
În 2018, Koselugo a fost desemnată de către FDA și EMA, „medicament orfan” pentru tratamentul pacienților pediatrici cu neurofibromatoză tip 1. În 2019, medicamentul a primit din partea FDA statutul de terapie „breakthrough“ în luna aprilie și „terapie pentru boală pediatrică rară” în decembrie. Astfel, la mai puțin de 6 luni, medicamentul este aprobat de către FDA și este așteptat ca în lunile următoare să urmeze și aprobarea din partea Agenției Europene a Medicamentului (EMA).
Citește și:
- #ASCO19. Modificările genetice care pot fi țintite prin terapii specifice în cancerele pediatrice sunt mai frecvente decât se considera anterior
- Boala Niemann-Pick: Alfa olipudaza, primul tratament care demonstrează rezultate pozitive, în studii clinice de fază avansată
- MiSight, primele lentile de contact care corectează și încetinesc progresia miopiei la copii, aprobate de FDA