
Trikafta, primul tratament indicat pentru 90% dintre pacienții cu fibroză chistică, aprobat de FDA
FDA a aprobat combinația elexacaftor/ivacaftor/tezacaftor, prima terapie indicată pacienților cu vârste de peste 12 ani cu fibroză chistică, ce prezintă măcar o mutație F508del, aceasta afectând aproximativ 90% dintre pacienți. Pacienții adolescenți cu fibroză chistică nu aveau până în acest moment altă opțiune terapeutică. FDA a luat această decizie cu 5 luni mai devreme decât data estimată pentru aprobare accelerată (martie 2020).

Trikafta a primit statutul prioritar (Priority Review) și pe cel de medicament inovator (Breakthrough Therapy) și a intrat în procedura accelerată de aprobare (Fast Track), trei programe care cresc accesul la terapiile sunt considerate a aduce adevărate schimbări de paradigmă. Trikafta este considerat și medicament orfan (Orphan Designation), devenind astfel, unul dintre puținele medicamente care au beneficiat de cele 4 mecanisme ale agenției americane.

Peste 2000 de mutații sunt asociate cu gena CFTR, iar dintre acestea F508del este cea mai frecventă. Majoritatea terapiilor disponibile până acum se adresau unor categorii restrânse de pacienți cu fibroză chistică.
Două studii clinice de fază 3 din programul AURORA au evaluat eficacitatea combinației terapeutice:
- 403 pacienți cu mutație F508del și o altă mutație pe care determină fie absența proteinei CFTR fie producerea unei proteine anormale, asociată cu ineficiența tratamentului cu ivacaftor în monoterapie sau tratamentului cu tezacaftor/ivacaftor
- 107 pacienți cu două mutații F508del identice.
Rezultatele ambelor studii sugerează că Trikafta ameliorează funcția pulmonară. Tratamentul a crescut volumul expirator maxim pe secundă (un parametru care evaluează progresia fibrozei chistice) cu aproape 14% în primul studiu și cu 10% în al doilea. De asemenea, pacienții au prezentat rate anuale de exacerbări mai scăzute și a avut efect pozitiv asupra indicelui de masă corporală comparativ cu placebo.
Datele sugerează creșterea speranței de viață pentru pacienții cu fibroză chistică. Dr. Elliot Dasenbrook, Respiratory Institute, Cleveland Clinic, Ohio a explicat:
„Rezultatul depinde și de momentul în care se administrează terapia. Pentru un copil de 12 ani acest medicament poate determina o durată a vieții normală. Aceasta este de fapt speranța. Chiar și pentru adulții cu un stadiu mai avansat al bolii tratamentul extinde supraviețuirea”
Profilul de siguranță a fost similar în toate subgrupurile analizate. Cele mai frecvente reacții adverse includ infecțiile respiratorii, cefaleea, creșterea enzimelor hepatice, rash-ul.
Cum se manifestă fibroza chistică?
Fibroza chistică este determinată de mutații la nivelul genei CFTR ( cystic fibrosis transmembrane conductance regulator), care codifică un canal transmembranar de clor de la nivelul celulelor glandelor exocrine, celule care produc mucus, enzime digestive sau saliva. Aparatul respirator și tractul gastrointestinal sunt cel mai frecvent implicate, însă în timp mai multe organe sunt afectate ceea ce conduce la complicații amenințătoare de viață.
Canalele ionice sunt structuri al cărui rol este permiterea mișcării atomilor sau moleculelor care au sarcină electrică. Pentru a ieși din celulă, ionii de clor trec prin acest canal special reprezentat de proteina CFTR. Transportul ionilor de clor permite controlul fluxului de apă în țesuturi, aceasta fiind necesară pentru asigurarea unei consistențe normale a mucusului. Mucusul e o substanță care protejează și lubrifiază căile respiratorii, sistemul digestiv, reproducător și alte organe și țesuturi.
La suprafața celulei ionii de clor atrag apa care permite mișcările cililor de la nivelul celulelor căilor respiratorii. Această mișcare determină și eliminarea mucusului. Atunci când proteina CFTR lipsește sau nu este funcțională clorul rămâne blocat în celule. Fără mișcarea ionilor de clor apa nu poate ajunge la suprafața celulară, iar mucusul blochează căile aeriene.
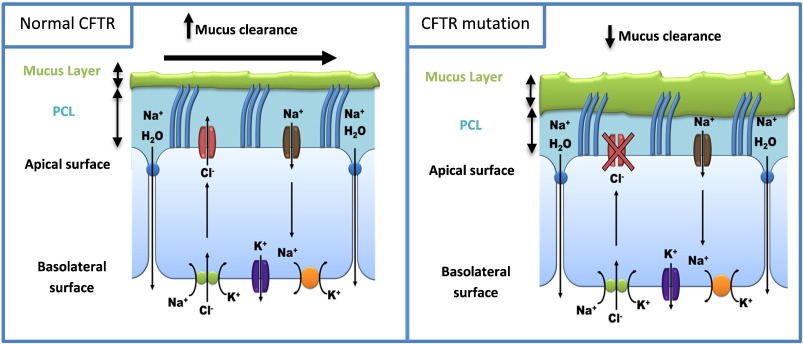
Mutațiile de la nivelul proteinei determină acumularea de mucus vâscos la nivelul căilor respiratorii și obstrucție, ceea ce conduce la infecții respiratorii grave. Infiltrația cu celule inflamatorii activează și mecanisme prin care apare distrucția țesuturilor.
La nivelul tractului gastro-intestinal apare afectarea canaliculelor pancreatice și ale vezicii biliare ceea ce împiedică fluxul de bilă și enzime în duoden și conduce la tulburări de absorbție a nutrienților. Apare, de asemenea, dezechilibru hidroelectrolitic din cauza pierderii de sodiu prin transpirație, cea ce conduce la deshidratare, aritmii, oboseală.
Tripla terapie aprobată de FDA face partea din categoria modulatorilor CFTR.
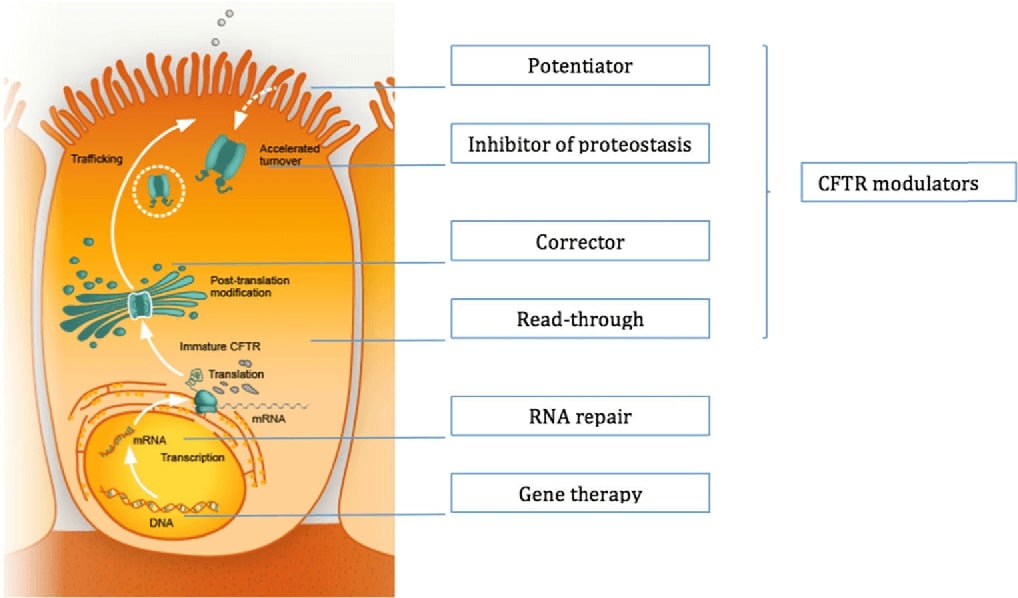
Există cel puțin 5 clase de mutații asociate bolii și peste 1900 de mutații identificate. Mutațiile de clasă I și II determină tulburări ale producției proteinei și se însoțesc de anomalii ale transferului proteinelor CFTR la suprafața celulei. În cazul mutațiilor de clasă III și IV, proteina care ajunge la suprafața celulară e de dimensiune normală însă există defecte ale activității de canal ionic. Mutațiile de clasă V afectează expresia de suprafață.
Ivacaftor a reprezentat primul tratament care și-a dovedit eficacitatea în studii clinice și a determinat creșterea activității proteinei CFTR, determinând ameliorarea VEMS, IMC și calității vieții pacienților. Acesta a fost aprobat în ianuarie 2012. Medicamentul se adresează unei mutații care apare în 5% dintre cazurile de FC. Ivacaftor crește fluxul de ioni prin canalul de clor activat de la suprafața celulară și este considerat un potențator CFTR.
Elexacaftor și tezacaftor sunt molecule care „corectează” proteina CFTR astfel încât aceasta să poată ajunge la locul corespunzător de la suprafața celulei. Ivacaftor permite deschiderea canalului de clor și facilitează pasajul clorului și sodiului.
9 din 10 pacienți cu fibroză chistică pot beneficia de tratament
„Această aprobare marchează un moment istoric prin care un nou tratament devine disponibil pentru majoritatea pacienților cu fibroză chistică, inclusiv adolescenții care anterior nu aveau o altă opțiune” – Ned Sharpless, comisarul FDA
Trikafta așteaptă în prezent evaluarea din partea Agenției Europene a medicamentului iar compania producătoare testează eficacitatea terapiei la pacienții pediatrici cu vârste între 6 și 8 ani.
Citește și:
- Genomica: știința avansează, dar populația și medicii sunt încă slab informați
- Dr. Olivier Tredan, despre testarea genomică de rutină
- ZIUA INTERNAȚIONALĂ A BOLILOR RARE 2018: Fibroza pulmonară idiopatică – 2 ani până la stabilirea diagnosticului corect, 54% dintre pacienți nu primesc tratamentul modern. De ce să aștepți?