
Sindromul VEXAS: o nouă boală genetică inflamatorie, cu implicare reumatologică și hematologică
Sindromul VEXAS, o boală rară identificată pentru prima dată în 2020, este mult mai frecvent decât s-a crezut inițial, potrivit cercetătorilor de la Universitatea din Leeds. Aceștia au dezvoltat un test genetic pentru a identifica pacienții care ar putea avea acest sindrom și vor să examineze mai multe persoane care prezintă simptome sugestive pentru a înțelege exact cât de frecventă este afecțiunea.
Sindromul VEXAS constă în febră fără o cauză identificabilă, erupții cutanate și afectare medulară. Bărbații mai în vârstă cu un sindrom autoinflamator nou instalat sunt adesea diagnosticați eronat cu policondrită recidivantă refractară la tratament, poliarterită nodoasă, sindrom Sweet sau arterită cu celule gigante. Conexiunea dintre aceste prezentări clinice diferite este reprezentată de mutații somatice la nivelul UBA1, o genă care inițiază un proces prin care proteinele pliate greșit sunt marcate pentru degradare. Sindromul VEXAS pare limitat la bărbați, deoarece gena UBA1 se află pe cromozomul X, deși poate fi întâlnit și la femei în cazul unei pierderi dobândite a cromozomului X. Mutațiile nu sunt prezente la naștere, ci sunt dobândite pe parcursul vieții.

VEXAS reprezintă un acronim pentru:
- Vacuole la nivelul măduvei osoase hematogene;
- Enzima activatoare E-1, codificată de gena UBA1;
- X-linkat;
- Autoinflamator;
- Mutație somatică cu mozaicism hematologic.
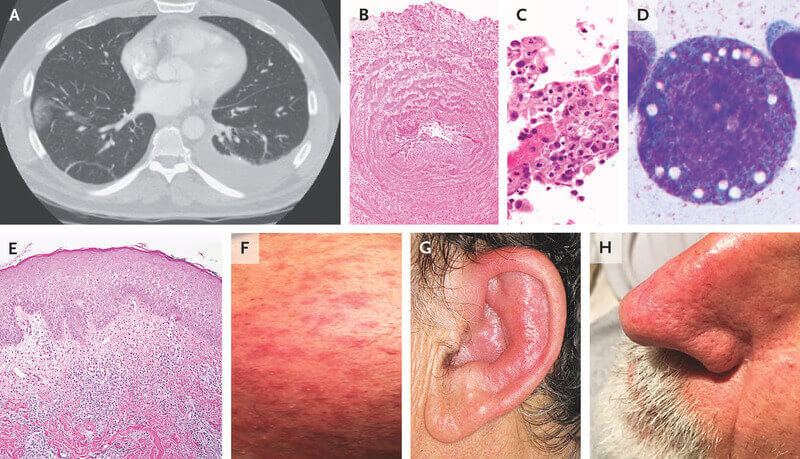
Afecțiunea a fost identificată prima dată în 2020. Investigațiile ulterioare conduse la Universitatea Leeds au identificat mutații genetice suplimentare care arată noi moduri în care boala se poate dezvolta. Echipa de cercetători a examinat probe de ADN pentru a stabili prevalența mutațiilor genetice identificate atunci când boala a fost descoperită. În cadrul ultimului studiu, a fost examinată o cohortă de 18 pacienți la care s-au identificat simptomele specifice și au fost identificate mutațiile la 10 dintre ei. Opt prezentau varianta genetică cunoscută anterior ca fiind asociată cu boala, însă doi pacienți aveau variante complet diferite. Astfel s-a identificat un nou mod în care mutațiile pot provoca sindromul VEXAS, ceea ce înseamnă că, probabil, numărul pacienților este mult mai mare decât se credea anterior. Prof. Dr. David B. Beck, unul dintre cercetătorii care au descoperit sindromul, a estimat că prevalența VEXAS ar putea fi de 1 la 20.000-30.000 de persoane.
La începutul lunii aprilie, alte 27 de cazuri fuseseră raportate, deoarece mai mulți medici au început să caute pacienții cu mutații ale genei UBA1, unii cu caracteristici clinice suplimentare celor deja cunoscute, inclusiv artrită inflamatorie cronică progresivă, boala Kikuchi-Fujimoto, spondiloartrită și pneumonie bacteriană.
În acest moment, cercetătorii investighează evoluția clinică naturală a pacienților identificați cu sindrom VEXAS, evaluând activitatea bolii și stabilind ce tratamente obțin un răspuns, cu scopul final al unui studiu terapeutic.
Majoritatea pacienților cu sindrom VEXAS au trecut printr-o mulțime de teste diagnostice, au încercat multe tratamente fără să reușească să obțină un răspuns. Acum, prin secvențierea ADN-ului pentru mutații ale genei implicate în apariția sindromului VEXAS, pacienții pot fi identificați și pot beneficia de cel mai bun tratament disponibil.
Un nou mod de a căuta boli genetice
Sindromul VEXAS a adus cu sine nu numai o nouă afecțiune medicală, ci și un nou mod în care cercetătorii pot descoperi o boală genetică. În loc să înceapă pe calea tradițională, căutând similitudini clinice în rândul pacienților cu boli nediagnosticate și apoi efectuând o căutare pentru o genă sau gene care ar putea explica simptomele comune, anchetatorii au schimbat abordarea, pornind de la genotip.
Aceștia au scanat secvențele genomice ale pacienților din rețeaua națională de boli nediagnosticate, ceea ce i-a condus la mutațiile genei UBA1 ca principal candidat. Au vizat calea ubiquitin-proteazom deoarece multe dintre bolile inflamatorii recurente sunt cauzate de mutații ale acestei căi. Apoi, au analizat genomul pacienților din alte baze de date naționale în căutarea mutațiilor somatice UBA1, identificând în cele din urmă 25 de bărbați cu caracteristici comune, pe care le-au denumit VEXAS. Aceste 25 de cazuri au constituit baza pentru raportul inițial despre acest sindrom publicat în revista științifică New England Journal of Medicine.
Majoritatea bolilor autoinflamatorii debutează în copilărie, deoarece provin din mutații ale liniei germinale. Sindromul VEXAS, din cauza mutațiilor somatice cu mozaicism, pare să se manifeste mai târziu în viață. Vârsta mediană a cohortei inițiale de 25 de bărbați a fost de 64 de ani, variind între 45 și 80 de ani. Mai mult decât atât, evoluția bolii pare a fi foarte severă. În momentul în care cercetătorii își pregăteau lucrarea pentru publicare, 10 din cei 25 de pacienți (40%) decedaseră.
„Cred că mutațiile somatice ar putea fi responsabile pentru un procent semnificativ de boli reumatologice severe cu debut la adulți, și ar putea schimba modul în care ne gândim la tratarea acestora pe baza diagnosticului genetic”, a declarat Dr. David B. Beck.
Cercetătorii consideră că un lucru important în acest studiu este că oferă un nou mod de a identifica boli: „Anterior căutam mutații care, în majoritatea cazurilor, erau aceleași mutații ale liniei germinale prezente la pacienți pediatrici care suferă de boli cu debutul timpuriu, dar acum am abordat lucrurile diferit. Poate exista un tip diferit de mutații care să conducă la boală reumatologică cu debut la vârsta adultă, iar acestea sunt mutații somatice care nu sunt prezente în fiecare celulă a organismului, decât în sânge”.
Când trebuie suspectat sindromul VEXAS?
Posibilitatea unui sindrom VEXAS ar trebui luată în considerare la bărbații de vârstă mijlocie sau mai în vârstă, cu caracteristici sugestive de policondrită recidivantă refractară la tratament, arterită cu celule uriașe, poliarterită nodoasă sau sindrom Sweet. Bărbații cu sindrom VEXAS prezintă adesea febră periodică, infiltrate pulmonare, antecedente de evenimente tromboembolice venoase neprovocate, dermatoze cu neutrofile și/sau anomalii hematologice, cum ar fi sindrom mielodisplazic, mielom multiplu sau gamapatie monoclonală de origine necunoscută. Biopsia măduvei osoase va identifica vacuole în celulele precursoare ale liniei mieloide și eritroide. Nivelurile markerilor inflamatori sunt foarte ridicate. Diagnosticul VEXAS poate fi confirmat numai prin teste genetice.
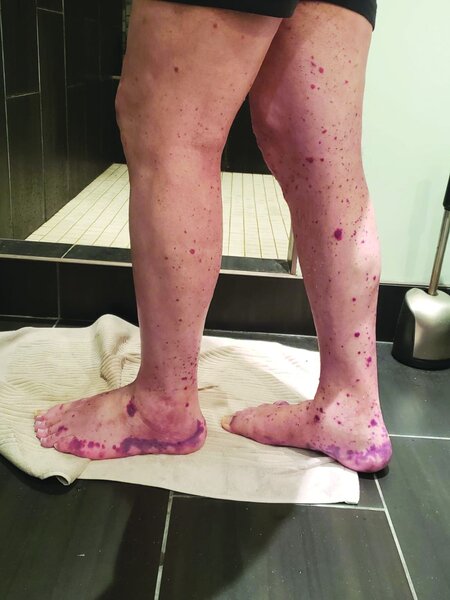
Managementul sindromului VEXAS necesită o echipă multidisciplinară de medici, inclusiv reumatologi, hematologi și dermatologi.
Un articol publicat recent în revista Arthritis & Rheumatology prezintă un algoritm clinic util pentru a decide dacă screeningul genetic pentru VEXAS este recomandat la pacienții cu policondrită recidivantă. Algoritmul menit să faciliteze identificarea VEXAS la pacienții care îndeplinesc criteriile de diagnostic pentru policondrita recidivantă se bazează pe câțiva factori ușor identificabili:
- sexul masculin;
- vârsta la debut peste 50 de ani;
- anemie macrocitară;
- trombocitopenie.
Conduita terapeutică
Dezvoltarea unor tratamente mai bune pentru sindromul VEXAS reprezintă o prioritate. În raportul inițial privind sindromul VEXAS, cercetătorii au descoperit că singura terapie eficientă sunt corticosteroizii în doze mari.
Reumatologul Marcela Ferrada, implicată în studierea sindromului, a spus că au început să se gândească la agenți care vizează atât caracteristicile hematologice, cât și cele inflamatorii ale VEXAS. “Majoritatea pacienților sunt expuși tratamentelor care vizează scăderea procesului inflamator și unele dintre aceste tratamente ajută parțial, dar nu complet, la scăderea cantității de corticosteroizi pe care pacienții le iau. De exemplu, unul dintre medicamente este tocilizumab, utilizat la pacienții care aveau un diagnostic anterior de policondrită recidivantă și primeau steroizi, însă manifestările lor hematologice continuau să progreseze. Suntem în curs de identificare a medicamentelor care pot ajuta la tratarea ambelor”. Ferrada a adăugat că, deoarece sursa mutației se află la nivelul măduvei osoase, transplantul medular ar putea fi o opțiune eficientă.
Cercetările pentru identificarea potențialelor tratamente pentru VEXAS ar putea identifica tratamente în afara agenților antiinflamatori clasici, cum ar fi direcționarea anumitor tipuri de celule din măduva osoasă sau calea ubiquitin-proteazomului, conform Dr. Beck. „Credem totuși că UBA1 acționează pentru a iniția inflamația, așa că poate fi importantă nu numai în VEXAS, ci și în alte boli.”
Citește și:
- Ziua mondială de luptă împotriva artritelor: pandemia COVID-19 a determinat digitalizarea accelerată a îngrijirii pacienților reumatologici
- STUDIU. Testele genetice adresate direct consumatorului determină în peste 80% din cazuri rezultate fals pozitive atunci când sunt folosite pentru identificarea mutațiilor rare