
Boala pulmonară interstițială determină 35% dintre decesele înregistrate la pacienții cu sclerodermie
Sclerodermia (scleroza sistemică) este o boală cronică rară, progresivă, autoimună, de cauză necunoscută. Se caracterizează prin fibroză difuză și anomalii vasculare la nivelul pielii, articulațiilor și organelor interne (mai ales tract gastro-intestinal, plămâni, inimă, rinichi). Afectarea pulmonară este frecventă și poate să apară în toate formele de boală.
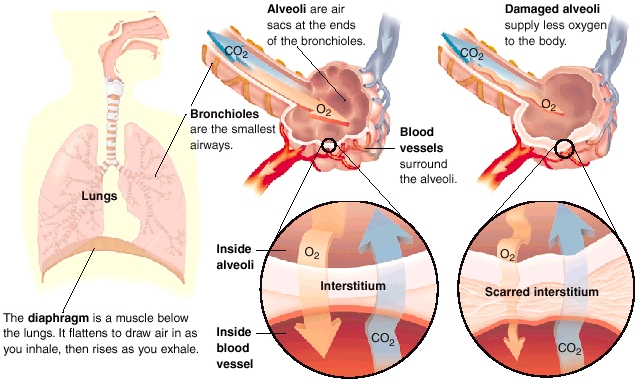
Modificările respiratorii din sclerodermie apar la nivelul întregului arbore respirator, inclusiv căi aeriene, parenchim, pleură, sistem vascular și muscular. Cea mai frecventă manifestare pulmonară a sclerodermiei este boala pulmonară interstițială – grup de boli pulmonare caracterizate de inflamația și îngroșarea (fibroza) interstițiului care înconjoară pereții alveolelor pulmonare. Se asociază cu modificări radiografice pulmonare specifice și cu prezența sindromului restrictiv la testele funcționale pulmonare.

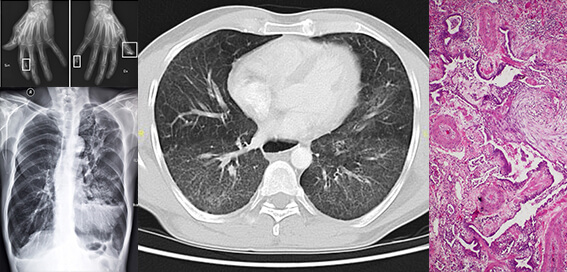
Incidența bolii pulmonare interstițiale asociată sclerodermiei variază în funcție de metoda de diagnostic. Testele funcționale pulmonare detectează defecte ventilatorii restrictive la 40-75% din pacienți, în timp ce tomografia computerizată cu rezoluție înaltă (HRCT) decelează modificări interstițiale la până la 90% dintre pacienți. Boala pulmonară interstițială este responsabilă de 35% din decesele pacienților cu sclerodermie, fiind principala cauză de mortalitate.
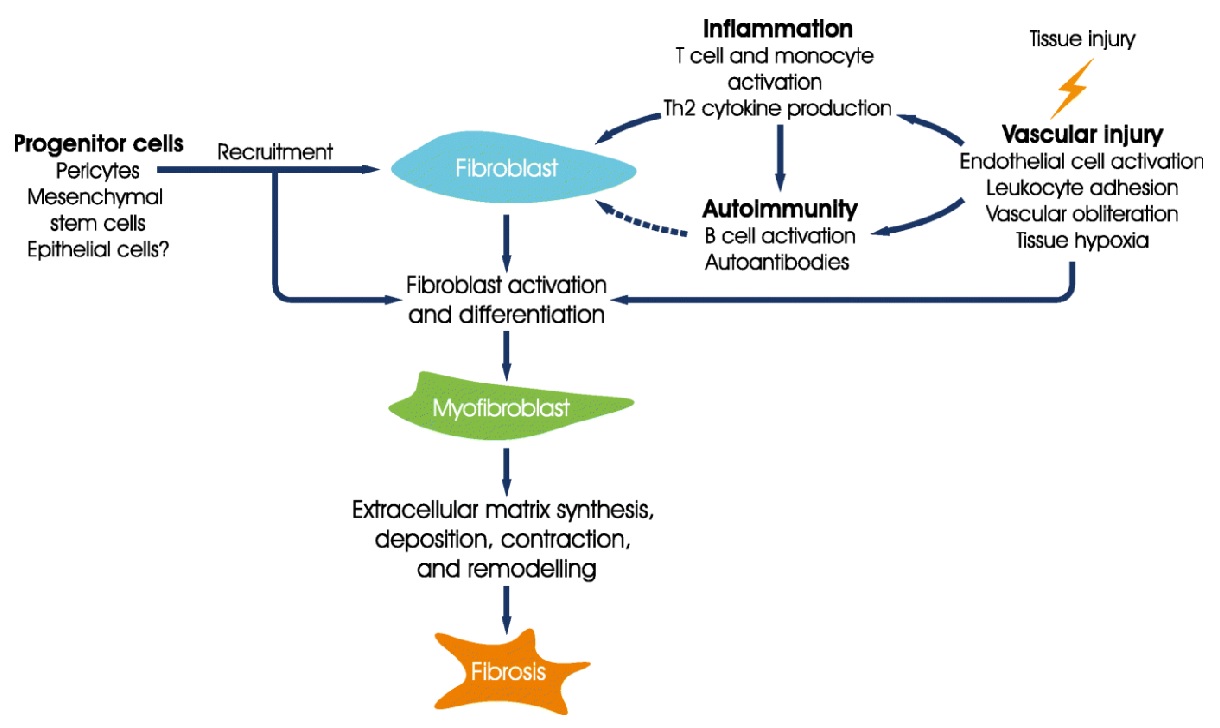
Patogeneza bolii pulmonare interstițiale asociată sclerodermiei a rămas necunoscută, în ciuda studiilor din ultimele decenii. Se consideră că este cauzată de relația complexă dintre imunitatea înnăscută și cea dobândită, inflamație, fibroză, care se produce la persoane cu susceptibilitate genetică; însă nu se cunoaște secvența desfășurării evenimentelor. Studii recente care au examinat profiluri de exprimare genică au oferit mai multe date despre patogeneza sclerodermiei. În viitor, asemenea informații moleculare și genetice pot oferi baza identificării unor biomarkeri predictivi, precum și dezvoltării de terapii țintite.
Manifestări clinice și paraclinice
Evoluția clinică a bolii pulmonare interstițiale variază foarte mult, de la formă ușoară, asimptomatică și stabilă, până la formă agresivă, rapid progresivă.
Cea mai frecventă modalitate de debut este dispnea de efort (respirația dificilă). Alte manifestări pot consta în: tuse neproductivă, cianoză, oboseală, scădere în greutate, durere în piept. La examenul clinic se decelează frecvent raluri uscate la bazele plămânilor. Mulți pacienți pot fi asimptomatici și să aibă examen clinic normal, motiv pentru care este important ca medicul să rămână vigilent și să continue urmărirea în evoluție pe parcursul bolii.

Principalele investigații utile în evaluarea afectării respiratorii sunt testele funcționale pulmonare, care ar trebui să includă cel puțin spirometria și capacitatea de difuziune a monoxidului de carbon (DLCO). Screening-ul trebuie realizat la toți pacienții în momentul stabilirii diagnosticului de sclerodermie deoarece modificările funcționale pot să apară înainte ca boala să devină clinic manifestă. Monitorizarea anuală a pacienților este un aspect crucial în managementul bolii pulmonare interstițiale. Modificările seriate sunt considerate semnificative dacă depășesc 10% între determinări în cazul CV, respect 15% în cazul DLCO.
Pacienții cu boală pulmonară interstițială asociată sclerodermiei au tulburare ventilatorie de tip restrictiv, semnalată prin scăderea capacității vitale (CV, sau FVC în engleză). Indicele Tiffeneau, de permeabilitate bronșică (raportul dintre volumul expirator mediu pe secundă și capacitatea vitală – VEMS/CV) este în general normal sau uneori puțin crescut, datorită scăderii VEMS în raport cu declinul CV. Inflamația parenchimatoasă și fibroza produc îngroșarea interstițială, ceea ce determină scăderea DLCO.
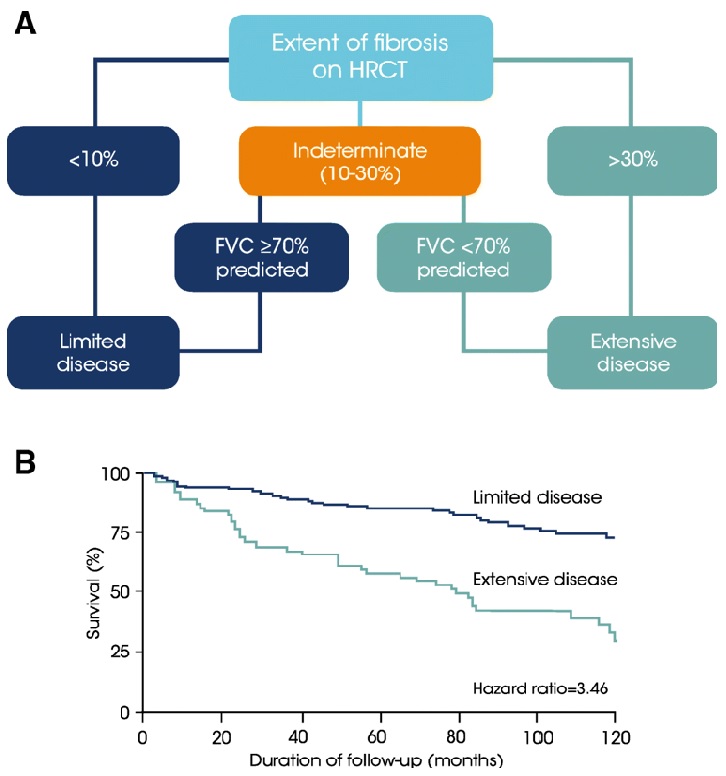
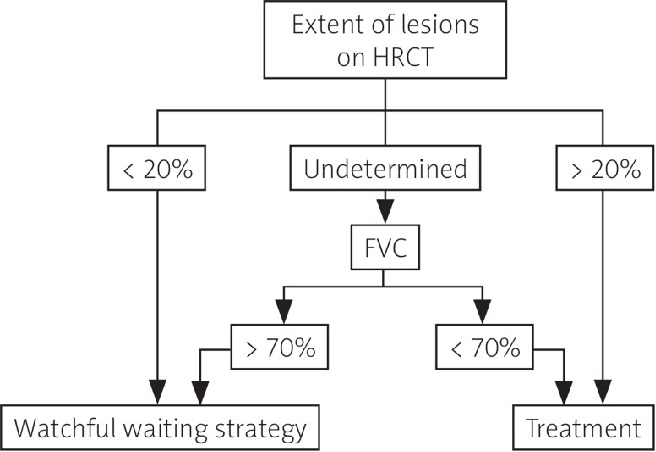
Tomografia computerizată cu rezoluție înaltă (HRCT) este cea mai utilizată investigație imagistică a bolii pulmonare. Gradul de extensie a fibrozei se corelează negativ cu CV și DLCO. Așadar, HRCT și testele funcționale pulmonare trebuiesc folosite în combinație pentru a evalua progresia și mortalitatea bolii pulmonare intersțiale asociată sclerodermiei.
Factori de risc pentru progresia bolii pulmonare interstițiale
Odată stabilit diagnosticul de sclerodermie, abordarea terapeutică trebuie individualizată fiecărui pacient: strategie de urmărire activă a evoluției sau începerea tratamentului imunosupresor. Urmărirea activă presupune monitorizare regulată prin teste funcționale pulmonare și investigații imagistice, urmată de începerea tratamentului atunci când se observă progresia bolii. Scopul este de a evita efectele adverse ale tratamentului dacă acesta nu este necesar și de a conserva calitatea vieții. Din acest motiv, este importantă prezicerea progresiei bolii pulmonare și identificarea factorilor de risc pentru progresie.
Principalii factori de risc pentru progresia bolii pulmonare interstițiale sunt:
- gradul modificărilor interstițiale vizibile la HRCT – fibroza care afectează > 20% din volumul plămânilor se asociază cu declin rapid al funcției pulmonare și cu mortalitate ridicată;
- gradul de declin al CV și al DLCO, mai ales în primii 5 ani de evoluție – predictori importanți ai evoluției spre boala pulmonară terminală;
- suprafața cutanată afectată – prevalența bolii pulmonare interstițiale este mai mare la pacienții cu scleroză sistemică generalizată (~50%), comparativ cu forma limitată (~35%);
- auto-anticorpii – 85% din pacienții care prezintă anticorpi anti-Scl-70 dezvoltă fibroză pulmonară, spre deosebire de cei care prezintă anticopir anti-centromer, care au risc mult mai mic de afectare pulmonară;
- nivelul plasmatic al CRP (proteina C reactivă – marker de inflamație);
- boala de reflux gastroesofagian asociată;
- prezența hipertensiunii arteriale pulmonare;
- vârsta avansată;
- sexul masculin – femeile au risc mai mare de a dezvolta sclerodermie, însă bărbații sunt mai predispuși de a prezenta forme cu evoluție severă.
Tratamentul bolii pulmonare interstițiale
Nu există criterii exacte pentru momentul inițierii tratamentului imunosupresor al bolii pulmonare interstițiale. Decizia este dificilă deoarece beneficiile terapeutice sunt modeste iar toxicitatea medicamentoasă este crescută. Din acest motiv, este importantă identificarea pacienților la risc de progresie și la care se impune începerea tratamentului, în funcție de criterii demografice, clinice, serologice și radiologice.
În ultimii 25 de ani, tratamentul imunosupresor a fost principala strategie terapeutică în boala pulmonară interstițială asociată sclerodermiei. Ciclofosfamida este singurul medicament care și-a dovedit eficacitatea și siguranța într-un studiu clinic multicentric, dublu-orb, controlat placebo (Scleroderma Lung Study I). Ciclofosfamida administrată oral timp de un an la pacienți cu boală pulmonară interstițială simptomatică a îmbunătățit funcția pulmonară (CV), capacitatea pulmonară totală, dispneea raportată de pacienți, scorul cutanat modificat Rodnan și mai mulți parametri ai calității vieții. Efectul benefic al ciclofosfamidei asupra CV s-a menținut la 18 luni de la inițierea tratamentului, însă a fost pierdut la 24 de luni.
Grupul EUSTAR (EULAR Scleroderma Trials and Research – bazat pe dovezi provenite din studii și pe consensul experților) recomandă tratamentul bolii pulmonare interstițiale asociată sclerodermiei cu ciclofosfamidă. Totuși, până la 1/3 din pacienți nu răspund la tratament și continuă să prezinte declin al funcției pulmonare. Din cauza eficacității limitate și a toxicității crescute a ciclofosfamidei, sunt necesare terapii imunosupresoare alternative.
Micofenolat mofetil este inhibitor al limfocitelor B și T. În cadrul unui studiu clinic randomizat (Scleroderma Lung Study II) s-a observat îmbunătățirea modestă și stabilizarea funcției pulmonare și a dispneei. Eficacitatea sa este comparabilă cu cea a ciclofosfamidei, însă micofenolat mofetil produce mai puține efecte adverse.
Azatioprina este o alternativă terapeutică la pacienții cu intoleranță sau contraindicații la ciclofosfamidă sau micofenolat.
Transplantul de celule stem hematopoietice este o opțiune de tratament la pacienți cu sclerodermie rapid progresivă și cu risc crescut de insuficiență de organ. Mortalitatea asociată tratamentului este ridicată din cauza riscului de infecții și alte complicații, astfel încât aceasta rămâne o alternativă limitată pentru o categorie restrânsă de pacienți.
O serie de agenți terapeutici, precum rituximab și tocilizumab, sunt evaluați momentan pentru posibilul efect benefic în tratarea bolii pulmonare interstițiale asociată sclerodermiei. Rezultatele preliminare sunt promițătoare însă sunt necesare studii ulterioare.