
FDA aprobă viltolarsen, a doua terapie țintită pentru pacienții cu distrofie musculară Duchenne, care acționează prin exon 53 skipping
Administrația Alimentelor și Medicamentelor din Statele Unite (FDA) a aprobat viltolarsen (denumire comercială Viltepso) pentru tratamentul pacienților cu distrofie musculară Duchenne (DMD), în cazul în care mutația cauzatoare poate fi tratată prin mecanisme de tipul exon 53 skipping. Se estimează că 8% dintre cazurile de DMD pot fi tratate prin acest mecanism.
“O prioritate a FDA este încurajarea dezvoltării de noi molecule contra bolilor neurologice grave, precum distrofia musculară Duchenne. Această aprobare aduce o opțiune terapeutică principală pentru pacienții a căror boală este cauzată de această mutație,” a afirmat Dr. Billy Dunn, directorul Departamentului de Neuroștiințe din cadrul FDA.
Viltolarsen a beneficiat de o serie de mecanisme FDA care accelerează aprobarea medicamentelor: aplicația a fost revizuită în mod prioritar (Priority review), deoarece această moleculă reprezintă un pas înainte important în tratamentul distrofiei Duchenne. Astfel, decizia de aprobare a fost luată în mai puțin de 6 luni de la depunerea aplicației, față de perioada standard de aproximativ 10 luni. În plus, datorită beneficiilor importante demonstrate de viltolarsen în studiile de fază II care stau la baza dosarului de aplicație, și aprobarea a fost emisă în mod accelerat (Accelerated Approval), iar molecula va fi evaluată în continuare în studii de fază III (studiul RACER53). În plus, viltolarsen a fost declarat de FDA Rare Pediatric Disease Drug și Orphan Drug.

Decizia de aprobare accelerată s-a bazat pe două studii de fază II: primul a inclus 16 pacienți cu vârsta de 4-10 ani, iar al doilea a inclus 16 pacienți cu vârsta de 5-18 ani. În primul studiu, pacienții au primit fie placebo, fie 40, fie 80 mg/kg corp/săptămână. Obiectivul principal a fost creșterea concentrației de distrofină din organism, proteina lipsă care cauzează degradarea musculară specifică bolii. Pacienții prezentau, la debutul studiului, valori de aprox. 0,6% din valorile normale pentru populația generală de distrofină, iar tratamentul a crescut aceste valori la 5,7% (pentru doza de 40 mg) și la 5,9% (doza de 80 mg). În plus, au fost ameliorați și parametri care cuantifică starea mușchilor din organism: viteza cu care pacientul se poate ridica din pat și durata de timp în care poate parcurge o distanță de 6 sau 10 metri. În al doilea studiu, concentrația distrofinei a crescut în mod semnificativ statistic doar în urma dozei de 80 mg/kg corp/săptămână.
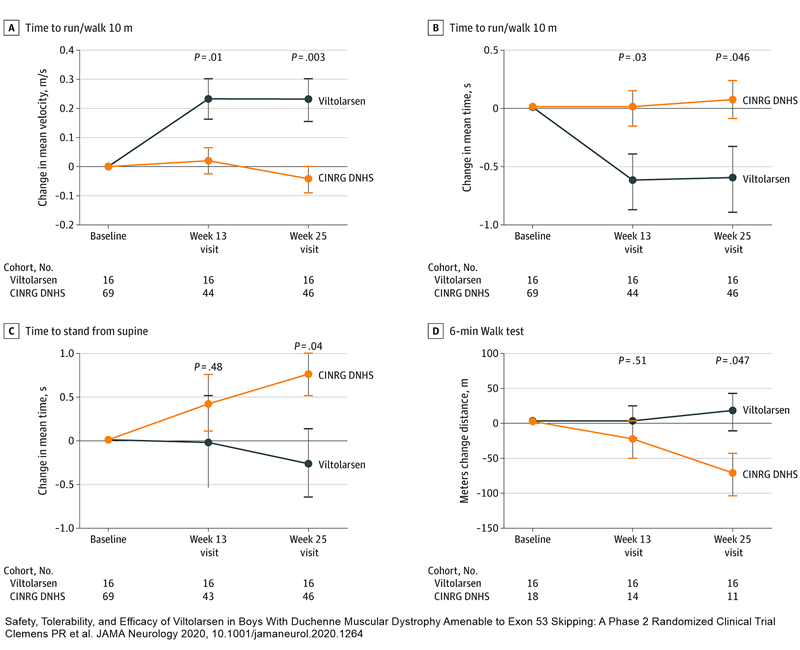
Cele mai frecvente reacții adverse raportate de studii sunt infecțiile de căi respiratorii superioare, tuse, febră sau inflamația regiunii în care se efectuează injecția. Nu au fost înregistrate reacții adverse severe, și niciun pacient nu a întrerupt tratamentul în urma reacțiilor adverse.
Despre distrofia Duchenne și tratamentele inovatoare recent aprobate
Distrofia musculară Duchenne este o boală genetică rară care implică deteriorarea progresivă a musculaturii scheletice. Simptomele apar din primii ani de viață, iar boala se poate agrava prin afectarea musculaturii cardiace și respiratorii. Având în vedere că defectul genetic cauzator se situează la nivelul cromozomului X, această boală afectează băieții (cu o incidență de 1:3.600 la nivel mondial), și extrem de rar și persoane de sex feminin.
Mecanismul care stă la baza acestei evoluții este absența proteinei distrofină, cu rol esențial în funcționarea și structura normală a celulei musculare striate. Gena care codifică structura proteinei este cea mai lungă din genomul uman (reprezentând 0,08% din totalul acestuia), fiind formată din 79 de exoni care stau la baza proteinei (exonul reprezentând unitate de bază a ADN-ului, care transcrie succesiuni de aminoacizi în cadrul proteinelor). Distrofia Duchenne poate fi cauzată de lipsa anumitor exoni, plasați în regiunile cheie ale genei: în lipsa acestora, producerea proteinei este întreruptă prematur și rezultă un lanț de aminoacizi mult prea scurt, nefuncțional.
O boală similară, cauzată de lipsa unui alt exon al distrofinei, este distrofia musculară Becker, în care exonul lipsă se află în regiuni mai puțin importante ale genei, iar în procesul de transcriere „se sare peste” exonul nefuncțional și rezultă o proteină cvasinormală, cu secvențe lipsă în regiuni neesențiale, însă cu structurile importante (capătul inițial și terminal) prezente. Astfel, distrofia Becker are manifestări clinice mai ușoare.
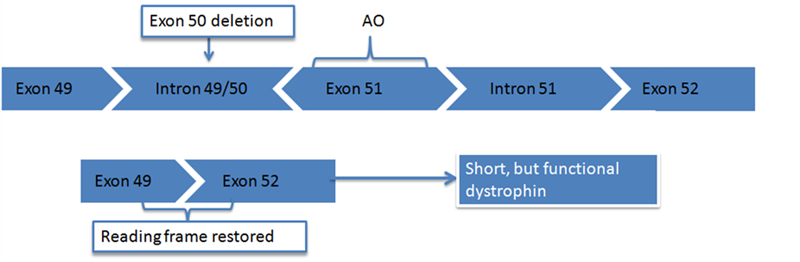
Având în vedere mecanismul genetic particular prin care apare distrofia Duchenne, o clasă terapeutică dezvoltată în ultimii ani s-a dovedit eficientă în controlul evoluției bolii: oligonucleotidele antisens. În procesul de producție al proteinei, exonii sunt transcriși, unul după altul, în succesiunea de aminoacizi normală. Oligonucleotidele antisens acționează prin legarea de exonul în discuție (în cazul viltolarsen, exonul 53 al distrofinei), împiedicând transcrierea acestuia în proteina finală. În cazul în care distrofia Duchenne este cauzată de lipsa unor exoni aflați anterior de exonul 53 (deleții ale exonilor 43–52, 45–52, 47–52, 48–52, 50–52 sau 52), inhibarea transcrierii exonului 53 atenuează efectul nociv al acestei lipse: în loc ca proteina să nu mai fie produsă deloc, transcrierea lanțului de exoni este finalizată, rezultând o proteină mai scurtă, însă parțial funcțională. Se estimează că aproximativ 8% dintre cazurile de
Alte oligonucleotide antisens sunt deja aprobate pentru tratamentul distrofiei Duchenne: eteplirsen, care acționează prin exon 51 skipping, golodirsen (exon 53 skipping). Niciuna dintre aceste molecule nu este aprobată de către EMA, pentru utilizare în UE. Pacienții din UE pot beneficia, totuși, de tratament cu ataluren, care acționează asupra ribozomilor (organitele intracelulare la nivelul cărora ARN-ul este transcris în proteine) și facilitează transcrierea distrofinei, în ciuda defectelor genetice.
Citește și:
- STUDIU. 1 din 17 pacienți cu boală cardiovasculară aterosclerotică prezintă o afecțiune genetică: hipercolesterolemie familială. În peste 90% dintre țările de pe glob nu se cunoaște prevalența HF
- Progrese în managementul distrofiei musculare Duchenne: FDA aprobă primul test de screening pentru nou-născuți și o nouă terapie țintită
- Zolgensma, prima terapie genică pentru atrofia musculară spinală, primește aprobare condiționată în UE