
Accesul la terapii inovatoare în Uniunea Europeană: sinteza activității EMA în al doilea trimestru al 2024
EMA (European Medicines Agency) este agenția Uniunii Europene responsabilă de evaluarea științifică, supravegherea și monitorizarea siguranței medicamentelor. În cadrul său funcționează o serie de comitete, rolul principal în autorizarea medicamentelor de uz uman revenind CHMP (Committee for Medicinal Products for Human Use).
CHMP se întrunește o dată pe lună pentru a evalua aplicațiile pentru avizarea introducerii pe piață, printr-o procedură centralizată, recomandând sau nu comercializarea fiecărui medicament analizat. Află mai multe: Anul 2023 din perspectiva accesului pacienţilor la terapii inovatoare în Uniunea Europeană. Care sunt medicamentele aprobate în primul trimestru de EMA?

Principalele noutăţi din cel de-al doilea trimestru al anului 2024:
- terapia genică Durveqtix pentru hemofilia B severă a fost aprobată
- noi opţiuni terapeutice pentru cancerele avansate au devenit disponibile în Uniunea Europeană
- tratament eficace pentru prevenţia hemoragiilor la pacienţii cu hemofilie A, aprobat de Comisia Europeană
- pentru prima dată, adrenalina este disponibilă sub formă de spray nazal pentru a fi administrată la persoanele cu reacţii alergice severe
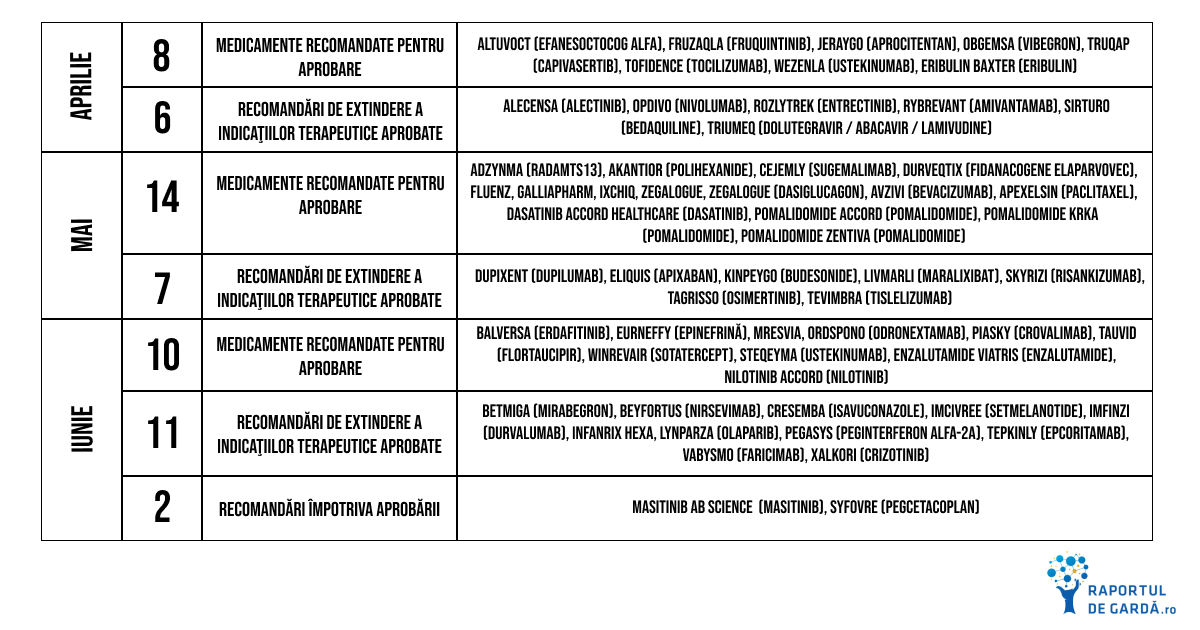
Trimestrul II din 2024 a debutat cu multe recomandări pozitive din partea CHMP
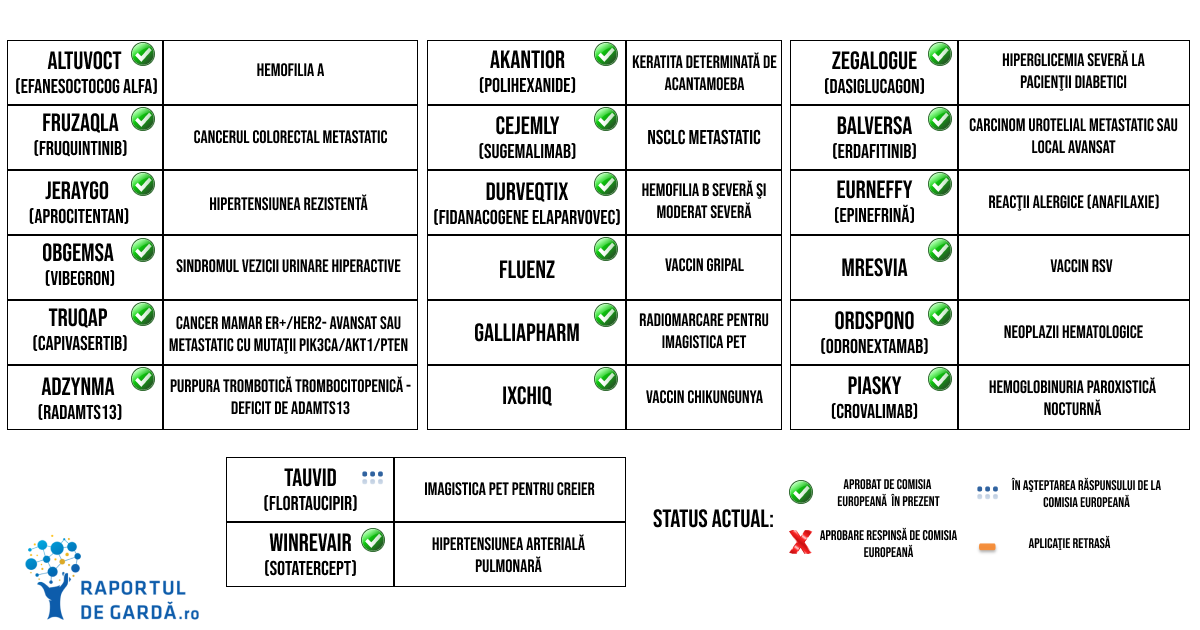
În aprilie, CHMP a oferit aviz pozitiv pentru Altuvoct, efanesoctocog alfa, cu indicaţia de terapie şi profilaxie a hemoragiilor la pacienţii cu hemofilie A. Această recomandare a fost urmată de aprobarea acordată de Comisia Europeană în luna iunie. Administrarea profilactică o dată pe săptămână a redus rata anuală a sângerărilor sub 1% şi peste 80% dintre pacienţi nu au prezentat hemoragii spontane, conform rezultatelor studiilor XTEND-1 şi XTEND-Kids.
Efanesoctocog alfa este o terapie de substituție a factorului VIII, conţinând un compus de fuziune, care include o regiune a factorului von Willebrand și polipeptide XTEN pentru a prelungi timpul de circulație. Este singura terapie care s-a dovedit a depăşi limitările legate de timpul de înjumătăţire a factorului von Willebrand, astfel că poate fi administrat o singură dată pe săptămână. Poţi afla detalii despre efanesoctocog alfa citind Profilaxia sângerărilor la copiii cu hemofilie A, eficientă și sigură cu efanesoctocog alfa, o singură doză pe săptămână.
Fruzaqla (fruquintinib), un inhibitor selectiv cu administrare orală a tuturor celor trei tipuri de receptori ai VEGF (vascular endothelial growth factor) a fost recomandat de CHMP pentru a fi utilizat ca monoterapie la pacienții adulți cu cancer colorectal metastatic care au fost tratați anterior cu terapiile standard disponibile, inclusiv chimioterapii pe bază de fluoropirimidină, oxaliplatină și irinotecan, agenți anti-VEGF și agenți anti-EGFR și a căror boală a progresat în cursul sau prezintă intoleranță la tratamentul fie cu trifluridină-tipiracil, fie cu regorafenib. Comisia Europeană a acordat autorizaţia în luna iunie cu această indicaţie.
Truqap (capivasertib) a fost aprobat în iunie în combinaţie cu Faslodex (fulvestrant), în urma avizului pozitiv acordat de CHMP în aprilie, pentru tratamentul pacienților adulți cu cancer de sân local avansat sau metastatic ER (receptori de estrogen) pozitiv, HER2 negativ, cu una sau mai multe modificări ale genelor PIK3CA, AKT1 sau PTEN, după recurență sau progresie în cursul sau după un regim bazat pe terapie endocrină.
Conform rezultatelor studiului CAPItello-291 , Truqap în asociere cu Faslodex a redus riscul de progresie a bolii sau deces cu 50% comparativ cu Faslodex în asociere cu placebo. Astfel, Truqap a devenit primul și singurul inhibitor AKT aprobat în Uniunea Europeană pentru pacienții cu cancer de sân cu modificări specifice ale biomarkerilor PIK3CA, AKT1 sau PTEN.
Poţi afla mai multe despre întâlnirea CHMP din aprilie aici.
De asemenea, în luna aprilie, un alt comitet din cadrul EMA, PRAC (Pharmacovigilance Risk Assessment Committee), a realizat o evaluare suplimentară a siguranţei a medicamentelor de tip agonist al receptorului GLP-1 (glucagon-like peptide-1), deoarece a fost ridicată problema asocierii acestor terapii cu gândurile suicidare şi de auto-vătămare.
Analiza condusă de PRAC a arătat că nu există nicio asociere de tip cauzal între administrarea agoniştilor GLP-1 şi tendinţele suicidare şi de auto-vătămare, concluzie foarte importantă, deoarece aceste medicamente devin din ce în ce mai populare pentru terapia diabetului zaharat de tip 2 şi, în cazul unor membri ai clasei, terapia obezităţii.
Acces la inovaţie în medicină, extins în mai 2024
CHMP a recomandat Durveqtix (fidanacogene elaparvovec) pentru autorizaţia condiţionată de comercializare în Uniunea Europeană, fapt urmat de aprobarea în luna iulie din partea Comisiei Europene. Durveqtix este o terapie genică adresată adulţilor cu hemofilie B severă şi moderat severă care nu prezintă inhibitori ai factorului IX (autoanticorpi anti-factor IX) şi care nu prezintă anticorpi detectabili împotriva virusului adenoasociat, serotipul Rh74 (AAVRh74var). În urma unei singure administrări, terapia stabileşte producţia de factor IX în corpul pacientului, astfel că sunt prevenite şi controlate sângerările.
Recomandarea are la bază rezultatele studiului clinic de fază III, care este încă în desfăşurare. Participanţii la studiu vor fi monitorizaţi timp de 15 ani. Studiul a inclus 45 de bărbaţi cu hemofilie B, fără anticorpi împotriva vectorului utilizat. A fost comparată rata anuală a sângerărilor (atât tratate, cât şi netratate) la pacienţii care au primit Durveqtix şi cei care au primit profilaxia de rutină cu factor IX. Rata anuală a sângerărilor a fost de sub 1,5 pentru Durveqtix, în timp ce pentru tratamentul standard a fost de 4,5.
Pe parcursul perioadei de monitorizare, care a variat de la 2 până la 4 ani, 60% dintre pacienţi nu au prezentat evenimente legate de sângerări, în comparaţie cu sub 30% în grupul care a primit factor IX. La pacienţii care au primit terapia genică, administrarea profilactică a factorului IX a fost redusă cu peste 90%. Reacţiile adverse constatate includ creşterea transaminazalor, care se normalizează prin administrarea corticosteroizilor, precum şi cefalee, simptome similare gripei, creşterea creatininei şi a lactat dehidrogenazei.
Cejemly (sugemalimab) a primit aviz pozitiv din partea CHMP în mai, recomandare urmată de aprobarea acordată de Comisia Europeană în luna iulie, pentru a fi administrat în combinaţie cu chimioterapia cu derivaţi de platină ca terapie de primă linie la pacienţii cu cancer pulmonar non-microcelular (NSCLC) metastatic, fără mutații acţionabile EGFR şi fără alteraţii ale genelor ALK, ROS1 sau RET. Mai multe despre Cejemly poţi afla citind articolul Imunoterapia sugemalimab, recomandată spre aprobare pentru cancerul pulmonar avansat în Uniunea Europeană.
Află detalii despre întâlnirea CHMP din mai.

Terapia Qalsody (tofersen), recomandată trimestrul trecut de CHMP, a fost aprobată în luna mai de Comisia Europeană pentru tratamentul unei forme genetice de scleroză laterală amiotrofică foarte rară, caracterizată printr-o mutație la nivelul genei pentru superoxid dismutaza 1 (SOD-1). Qalsody este o terapie de tip oligonucleotid antisens, care se atașează de ARN-ul mesager SOD1, limitând producția proteinei. Conform rezultatelor studiului clinic de fază 3 VALOR, la pacienții care au primit tofersen a fost constatată scăderea nivelurilor plasmatice ale lanțului ușor al neurofilamentelor, marker pentru leziuni ale axonilor neuronali și nerodegenerare.
În cadrul întâlnirii PRAC din mai, s-a concluzionat că pacienţii care primesc terapii CAR-T trebuie monitorizaţi pentru tot restul vieţii pentru a depista eventuale malignităţi secundare ale limfocitelor T, deoarece această metodă de tratament creşte riscul de dezvoltarea a unui nou cancer, diferit de cel pentru care a fost administrată terapia CAR-T. Dezvoltarea cancerelor secundare este foarte rară la pacienţii care au primit terapii CAR-T, însă s-a demonstrat că celulele CAR-T sunt implicate direct în procesul de malignizare.
Veşti bune din partea CHMP şi în luna iunie
Balversa (erdafitinib) a primit aviz pozitiv din partea CHMP ca monoterapie cu administrare orală pentru tratamentul pacienților adulți cu carcinom urotelial nerezecabil sau metastatic, care prezintă modificări genetice FGFR3 susceptibile la terapie, care au primit anterior cel puțin o linie de tratament care conține un inhibitor PD-1 sau PD-L1 în contextul bolii nerezecabile sau metastatice. Balversa este un inhibitor pan-FGFR, devenind în august prima terapie de acest tip disponibilă în Uniunea Europeană cu această indicaţie.
Conform rezultatelor studiului THOR, pacienţii care au primit erdafitinib au avut o supravieţuire generală mediană cu peste 4 luni mai îndelungată în comparaţie cu cei care au primit chimioterapie (12.1 luni versus 7.8 luni). Noua terapie a determinat, de asemenea, dublarea supravieţuirii în lipsa progresiei în comparaţie cu chimioterapia (5.6 luni versus 2.7 luni).
Urmând recomandării pozitive a CHMP din iunie, Comisia Europeană a aprobat Ordspono (odronextamab) pentru tratamentul pacienților adulți cu limfom folicular recidivat sau refractar sau limfom difuz cu celule B mari recidivat sau refractar, după două sau mai multe linii de terapie sistemică. Aceasta marchează prima aprobare la nivel mondial a Ordspono pentru această categorie de pacienți. Ordspono este un anticorp bispecific, care acționează prin conectarea celulor maligne cu limfocitele T, facilitând activarea acestora şi eliminarea cu succes a celulelor neoplazice.
La pacienţii cu limfom folicular recidivat sau refractar, rezultatele studiului ELM-2 au indicat pentru odronextamab o rată de răspuns obiectiv de 80%, 73% obținând un răspuns complet. La persoanele la care a fost obţinut răspunsul complet, durata medie a răspunsului a fost de 25 de luni.
La pacienţii cu limfom difuz cu celule B mari recidivat sau refractar care nu au primit terapii CAR-T, rezultatele de la ELM-2 au indicat o rată de răspuns obiectiv de 52%, 31% obţinând răspuns complet, cu o durată mediană a răspunsului de 18 luni. La pacienţii cu limfom difuz cu celule B mari recidivat sau refractar care au progresat după terapia CAR-T, rezultatele studiului ELM-1 au indicat o rată de răspuns obiectiv de 48%, 32% obţinând remisiunea completă, cu o durată mediană a răspunsului de 15 luni.
Tot în luna iunie, CHMP a oferit aviz pozitiv pentru EURneffy, primul spray pentru administrare nazală a adrenalinei la pacienţii cu şoc anafilactic şi alte reacţii alergice. Recomandarea a fost urmată de aprobarea din partea Comisiei Europene în luna august. Disponibilitatea unui preparat de acest tip, cu administrare nazală are un impact foarte important. Administrarea injectabilă a adrenalinei în cazul episoadelor severe de reacţii alergice a salvat numeroase vieţi, însă au existat cazuri când pacientul sau persoanele din jur au ezitat să utilizeze epinefrina din teamă. Administrarea nazală este mult mai convenabilă şi simplă pentru oricine, fie că este vorba de autoadministrare sau nu.
De asemenea, CHMP a recomandat în iunie aprobarea Winrevair (sotatercept), un nou tip de terapie biologică pentru hipertensiunea pulmonară, afecţiune cu prognostic nefavorabil, jumătate dintre pacienţi decedând în 5-7 ani de la diagnostic. Acest tratament prezintă un nou mecanism de acțiune, fiind primul inhibitor al căii de semnalizare prin activina pentru pacienții cu hipertensiune pulmonară. Comisia Europeană a autorizat în august comercializarea Winrevair. Poţi afla detalii despre această nouă terapie citind FDA aprobă sotatercept, un nou tip de terapie biologică pentru hipertensiunea arterială pulmonară.
Află mai multe despre întâlnirea CHMP din iunie.
Citeşte şi:
- Accesul la terapii inovatoare în Uniunea Europeană: sinteza activității EMA în primul trimestru al 2024
- Anul 2023 din perspectiva accesului pacienţilor la terapii inovatoare în Uniunea Europeană. Care sunt medicamentele aprobate în al treilea trimestru de EMA
- Anul 2023 din perspectiva accesului pacienţilor la terapii inovatoare în Uniunea Europeană. Care sunt medicamentele aprobate în ultimul trimestru de EMA