
Anul 2023 din perspectiva accesului pacienţilor la terapii inovatoare în Uniunea Europeană. Care sunt medicamentele aprobate în primul trimestru de EMA?
La finalul anului 2023, Raportuldegardă.ro vă prezintă principalele progrese înregistrate în domeniul terapiilor inovatoare în Uniunea Europeană (UE) în ultimele 12 luni, medicamentele disponibile sau care au primit aviz pozitiv pentru aprobare în UE, precum și rezultatele studiilor care au constituit baza autorizării pentru comercializare. Cele mai importante realizări din primul trimestru al anului 2023:
- aprobarea terapiei genice Hemgenix pentru hemofilia B de către Comisia Europeană
- prima terapie de tip inhibitor selectiv TYK2 a fost aprobată, fiind adresată pacienților cu psoriazis în plăci
- prima terapie ţintită a fost aprobată pentru esofagita eozinofilică, precum și pentru dermatita atopică la copii mai mici de 5 ani
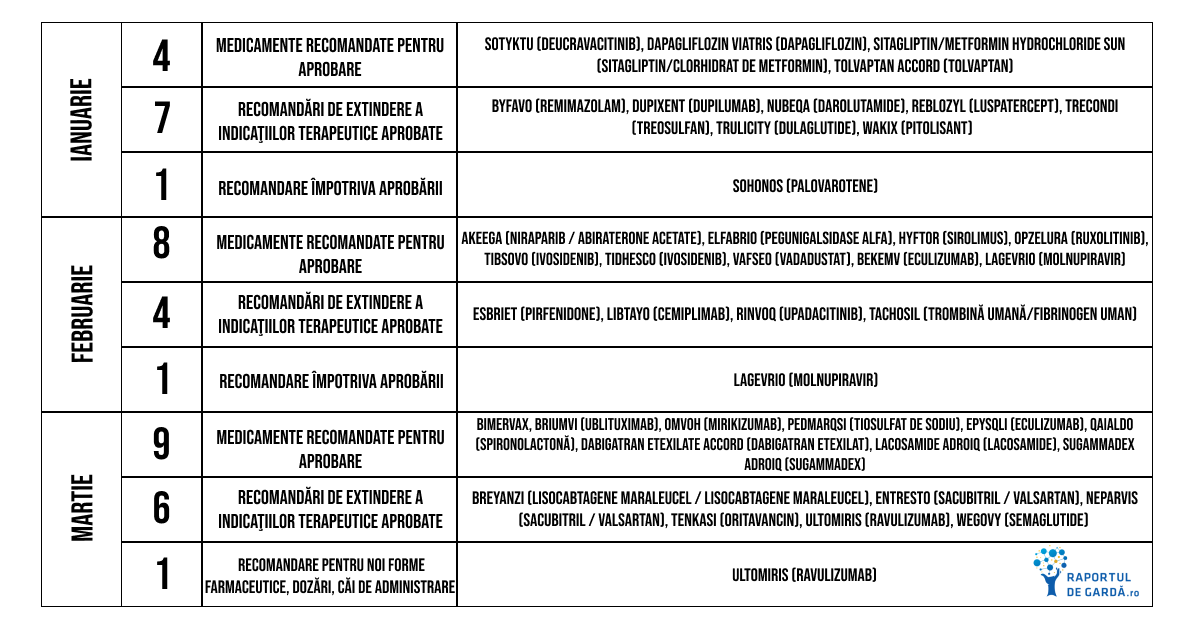
Principalele noutăţi din luna ianuarie 2023
În urma rezultatelor studiului clinic de fază III DESTINY-Breast04, Enhertu (trastuzumab deruxtecan) a fost aprobat în Uniunea Europeană pentru monoterapia adulţilor cu cancer de sân nerezecabil sau metastatic, cu expresie scăzută a HER2 (HER2-low), care au primit anterior chimioterapie în context metastatic sau care au prezentat recurenţe în timpul sau în următoarele 6 luni de la finalizarea chimioterapiei adjuvante. Obţinerea acestei aprobări a oferit, pentru prima dată, oportunitatea de a trata cu o terapie ţintită HER2 pacienţii care au niveluri scăzute ale expresiei acestui marker.

Enhertu este o terapie de tip ADC (antibody-drug conjugate), care a redus riscul de progresie a bolii sau deces cu 50%, în comparaţie cu chimioterapia la pacienţii cancer de sân metastatic HER2-low, pozitiv sau negativ pentru receptorul hormonal. Mediana PFS (progression-free survival) a fost aproape dublă în urma terapiei cu Enhertu, în comparaţie cu chimioterapia (aproximativ 10 luni vs. puţin peste 5 luni). Mediana OS (overall survival) a fost de peste 23 luni la pacienţii care au primit Enhertu, în timp ce la cei care au primit chimioterapie, a fost de aproape 17 luni.
În aceeași lună, terapia de tip anticorp monoclonal Dupixent (dupilumab) a primit aprobarea din partea Comisiei Europene pentru a fi administrată pacienţilor cu esofagită eozinofilică. Rezultatele studiului de fază III au indicat o rată de 10 ori mai ridicată (aproximativ 60%) de obţinere a remisiei histologice, în comparaţie cu placebo (aproximativ 6%). Simptomele au fost reduse în proporţie de 60-70% în urma administrării Dupixent, iar îmbunătăţirea stării clinice a fost aproape dublă faţă de placebo.
Dupixent reprezintă prima şi singura terapie ţintită disponibilă pentru pacienţii cu esofagită eozinofilică. Mecanismul său de acţiune constă în inhibiţia semnalizării prin intermediul căilor interleukinelor 3 şi 13, fără a prezenta efecte de tip imunosupresor.
În luna ianuarie, în cadrul întâlnirii lunare CHMP, a fost oferit avizul pozitiv pentru noua terapie Sotyktu (deucravacitinib), indicată adulţilor cu psoriazis în plăci, de tip moderat-sever. Detalii despre întâlnirea lunară CHMP din ianuarie aici.
Inovațiile care au devenit disponibile în februarie 2023 în Uniunea Europeană
Principala noutate a lunii februarie a fost reprezentată de aprobarea Hemgenix (etranacogene dezaparvovec) de către Comisia Europeană, adresată hemofiliei B severe şi moderat-severe, la adulţii fără istoric de inhibitori ai factorului IX al coagulării. Hemgenix a devenit, astfel, prima terapie genică aprobată pentru Hemofilia B în Uniunea Europeană şi Spaţiul Economic European.
În cadrul studiului clinic HOPE-B, s-a demonstrat că administrarea Hemgenix creşte nivelul activităţii factorului IX al coagularii în mod stabil şi durabil, reducând rata anuală a sângerărilor cu aproape 65%. Utilizarea profilactică a factorului IX a fost întreruptă la 96% dintre pacienţii incluşi în studiu, iar consumul factorului IX al coagulării a fost redus cu 97% la 18 luni de la administrarea Hemgenix.
Hemgenix presupune o singură administrare a variantei Padua a genei pentru factorul IX, prin intermediul unui virus adeno-asociat (AAV5). Această genă codifică un factor IX de 5-8 ori mai activ decât cel sintetizat pe baza genei comune.
Combinaţiile de imunoterapii în oncologie
Tot în luna februarie, Comisia Europeană a aprobat administrarea în combinaţie a imunoterapiilor Imjudo (tremelimumab) şi Imfinzi (durvalumab) pentru tratamentul de primă linie al adulţilor cu carcinom hepatocelular (HCC) avansat sau nerezecabil, precum şi utilizarea acestora împreună cu chimioterapia cu derivaţi de platină la pacienţii adulţi cu cancer pulmonar non-microcelular (NSCLC) metastatic (stadiul IV). Aprobarea a fost realizată pe baza rezultatelor studiilor clinice de fază III HIMALAYA şi POSEIDON.
Studiul HIMALAYA a arătat că terapia combinată a redus riscul de deces cu 22% la pacienţii cu HCC, în comparaţie cu administrarea sorafenib. Numărul de pacienţi care au supravieţuit minim 3 ani a fost cu peste 10% mai ridicat în grupul care a primit Imjudo şi Imfinzi, faţă de utilizarea sorafenibului.
Cele două imunoterapii, utilizate împreună cu chimioterapia cu derivaţi de platină, au redus cu 25% riscul de deces şi cu 28% riscul de progresie, în comparaţie cu chimioterapia, conform rezultatelor studiului POSEIDON. De asemenea, numărul de pacienţi care au supravieţuit minim 3 ani a fost cu peste 10% mai ridicat în urma administrării regimului terapeutic combinat, faţă de chimioterapie.
Imfinzi (durvalumab) este un anticorp monoclonal uman care se ataşează de proteina PD-L1 şi în împiedică interacţiunea cu PD-1 şi CD-80. Astfel, întrerupe căile prin care celulele maligne evită să fie eliminate de sistemul imun. Imjudo (tremelimumab) este un anticorp uman monoclonal care ţinteşte CTLA-1 (cytotoxic T-lymphocyte- associated protein 4), contribuind la activarea limfocitelor T și eliminarea celulelor neoplazice.

În luna februarie, în cadrul întâlnirii lunare CHMP, a fost oferit avizul pozitiv pentru noile terapii Akeega (niraparib / abiraterone acetate), Elfabrio (pegunigalsidase alfa), Hyftor (sirolimus), Opzelura (ruxolitinib), Tibsovo (ivosidenib), Tidhesco (ivosidenib), Vafseo (vadadustat). Detalii despre întâlnirea lunară CHMP din februarie aici.
Ce noutăți au marcat luna martie 2023?
Sotyktu (deucravacitinib) a fost aprobat de Comisia Europeană pentru tratamentul adulţilor cu psoriazis în plăci moderat spre sever, care sunt candidaţi pentru terapie sistemică. Sotyktu a devenit astfel, primul inhibitor TYK2 aprobat, indiferent de indicaţie.
Sotyktu ţinteşte selectiv TYK2, inhibând semnalizarea realizată de interleukinele 23, 12 şi de interferonul de tip 1, citokine implicate în patologia a multiple afecţiuni cu substrat imunologic. La doze terapeutice, nu inhibă kinazele Janus (JAK) 1, 2, 3. Aprobarea a fost fondată pe rezultatele studiilor clinice de fază III POETYK PSO-1 şi POETYK PSO-2. Eficacitatea Sotyktu a fost superioară în comparaţie cu placebo şi apremilast.
Tot în luna martie, o nouă indicaţie terapeutică a fost aprobată de către Comisia Europeană pentru Dupixent (dupilumab): tratamentul dermatitei atopice severe la copiii cu vârste între 6 luni şi 5 ani, care sunt candidaţi pentru terapie sistemică. Aproape de 7 ori mai mulţi copii care au primit Dupixent au prezentat remiterea completă sau aproape completă a leziunilor cutanate şi reducerea severităţii generale a bolii, în comparaţie cu placebo.
Libtayo (cemiplimab), în combinaţie cu chimioterapia cu derivaţi de platină, a fost aprobat, de asemenea, de Comisia Europeană pentru tratamentul de primă linie al pacienţilor adulţi cu NSCLC avansat cu expresie PD-L1 peste 1%, incluzând pacienţii care nu au mutaţii EGFR, ALK sau ROS1 şi ale căror tumori sunt metastatice sau local avansate şi care nu sunt candidaţi pentru chemoradiere definitivă. Libtayo este un anticorp monoclonal uman direcţionat împotriva receptorului PD-1 de pe suprafaţa limfocitelor T. Acesta previne utilizarea căii PD-1 de către celulele maligne pentru a inhiba activarea limfocitelor T.
În luna martie, în cadrul întâlnirii lunare CHMP, a fost oferit avizul pozitiv pentru noile terapii Bimervax, Briumvi (ublituximab), Omvoh (mirikizumab), Pedmarqsi (tiosulfat de sodiu). Detalii despre întâlnirea lunară CHMP din martie aici.
Autorizarea medicamentelor în Uniunea Europeană. Cum funcționează?
EMA (European Medicines Agency) reprezintă o agenție a Uniunii Europene, responsabilă de evaluarea științifică, supravegherea și monitorizarea siguranței medicamentelor. În cadrul său funcționează o serie de comitete, rolul principal în autorizarea medicamentelor de uz uman revenind CHMP (Committee for Medicinal Products for Human Use).
CHMP se întrunește o dată pe lună pentru a evalua aplicațiile pentru avizarea introducerii pe piață, printr-o procedură centralizată. Lunar, EMA publică o listă cu medicamentele pentru uz uman care sunt în curs de analizare de către CHMP. În urma evaluării științifice a fiecărei aplicații, CHMP recomandă sau nu comercializarea fiecărui medicament.
Pentru produsele medicamentoase de tip avansat (advanced therapy medicinal products, ATMPs), care includ terapiile genice, terapiile celulare de tip somatic și terapiile obținute prin bioingineria țesuturilor, există un comitet suplimentar – CAT (Committee for Advanced Therapies). CAT asigură expertiza necesară pentru a evalua aplicațiile de comercializare pentru ATMPs, emițând opinii cu privire la calitatea, siguranța și eficacitatea acestora. CAT trimite rezultatul evaluării către CHMP, pe baza căruia CHMP va recomanda sau nu autorizarea de către Comisia Europeană.
Procedura centralizată de autorizare presupune transmiterea unei singure aplicații de către companiile farmaceutice către EMA. Obținerea aprobării pentru comercializare permite inserția medicamentului pe piață și disponibilitatea acestuia pentru pacienții din Uniunea Europeană, pe baza unei singure autorizații. Conform reglementărilor Uniunii Europene, EMA nu are autoritatea de a permite comercializarea medicamentelor.
Comisia Europeană este autoritatea care poate autoriza comercializarea medicamentelor, pe baza recomandărilor EMA, în aproximativ 2 luni de la recepționarea acestora. Odată emisă autorizația, aceasta este valabilă în toate statele membre ale Uniunii Europene și în țările din Spațiul Economic European Iceland, Liechtenstein și Norvegia. Însă, înainte ca un medicament să devină disponibil într-una dintre aceste state, deciziile referitoare la preț și rambursare sunt adoptate la nivel național sau regional.

Autorizarea medicamentelor poate fi condiționată sau ”definitivă”. În interesul sănătății publice, se poate acorda o autorizație de introducere pe piață condiționată pentru anumite medicamente pe baza unor date clinice mai puțin complete decât cele necesare în mod normal, în cazul în care beneficiul disponibilității imediate a medicamentului depășește riscul inerent faptului că sunt încă necesare date suplimentare.
Exemple de medicamente de uz uman eligibile pentru a primi autorizație de introducere condiționată pe piață sunt cele care sunt destinate tratării, prevenirii sau diagnosticării unor boli grave debilitante sau care pun viața în pericol, celor necesare în cazul urgențelor de sănătate publică (de exemplu, pandemia COVID-19). Autorizația de comercializare completă este oferită în contextul recepționării și evaluării tuturor datelor necesare pentru a stabili eficacitatea și siguranța unui medicament.
Companiile farmaceutice pot depune cereri de autorizare pentru comercializare pentru medicamente noi, indiferent că sunt originale sau generice, biosimilare, de tip hibrid sau prin consimțământ informat, precum și cereri de autorizare a extinderii indicațiilor pentru medicamente deja comercializate în UE.
Farmacologie 101 – dicționar specific (conform EMA)
Aplicație pentru autorizarea unui medicament de tip consimțământ informat: o cerere în care deținătorul autorizației de comercializare a medicamentului de referință și-a dat consimțământul pentru utilizarea datelor medicamentului de referință pentru cerere.
Medicament biologic: un medicament a cărui substanță activă este produsă de un organism viu.
Medicament biosimilar: un medicament care este similar cu un medicament biologic care a fost deja autorizat.
Medicament generic: un medicament dezvoltat pentru a fi identic cu un alt medicament de marcă (brand-name) deja comercializat în ceea ce privește forma de dozare, siguranța, concentrația, calea de administrare, calitatea, caracteristicile de performanță și utilizarea prevăzută. Aceste asemănări contribuie la demonstrarea bioechivalenței, ceea ce înseamnă că un medicament generic funcționează în același mod și oferă același beneficiu clinic ca și medicamentul de marcă. O companie poate comercializa un medicament generic numai după ce perioada de exclusivitate de 10 ani pentru medicamentul original a expirat.
Medicament hibrid: un medicament care este similar cu un medicament autorizat care conține aceeași substanță activă, dar în care există anumite diferențe între cele două medicamente, cum ar fi concentrația, indicația sau forma farmaceutică.
Medicament inovator: un medicament care conține o substanță activă sau o combinație de substanțe active care nu au fost autorizate anterior.
Medicament de tip orfan: un medicament pentru diagnosticarea, prevenirea sau tratamentul unei afecțiuni care pune viața în pericol sau este debilitantă în mod cronic, care este rară (afectează maxim 5/10.000 de persoane din UE) sau situațiile în care medicamentul este puțin probabil să genereze profit suficient pentru a justifica cercetarea și dezvoltarea cheltuieli.
Medicament original (brand): medicament nou care a fost înregistrat și comercializat pentru prima dată.
Medicament personalizat: un medicament care se adresează pacienților individuali, pe baza caracteristicilor genetice ale acestora.
Substanță activă: substanța responsabilă de activitatea unui medicament.
Produs medicamentos: substanță sau combinație de substanțe care au scopul de a trata, preveni sau diagnostica o boală sau de a restabili, corecta sau modifica funcții fiziologice prin exercitarea unei acțiuni farmacologice, imunologice sau metabolice.
Produs medicamentos investigațional: un medicament care este evaluat în cadrul unui studiu clinic.
Citește și:
- Criza de medicamente din pandemie schimbă politica UE. Care sunt măsurile Comisiei Europene
- UE reformează legislația farmaceutică. Către cine înclină balanța beneficiilor?
- EMA facilitează implicarea grupurilor de pacienți în procesul de evaluare a noilor medicamente încă de la început